2
Pharmacokinetics
This chapter will be most useful after having a basic understanding of the material in Chapter 2, Pharmacokinetics in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. This chapter also draws from content in Chapter 5, Membrane Transporters and Drug Response; Chapter 6, Drug Metabolism; and parts of Chapter 7, Pharmacogenetics. The drugs presented in this chapter are used to illustrate general pharmacokinetic principles. The pharmacokinetic, pharmacodynamic, and therapeutic uses of drugs described in Chapter 2 are discussed in more detail in subsequent chapters. Neither a Mechanisms of Action Table nor a Clinical Summary Table is included in this chapter because this information is provided in subsequent chapters.
In addition to the material presented here, the 12th Edition contains:
• A review in Chapter 2 of cell membranes and their physicochemical properties that affect the movement of drugs to their site of action
• A discussion in Chapter 2 of different routes of drug administration and their relative advantages and disadvantages
• A detailed discussion in Chapter 2 of renal drug excretion, biliary and fecal excretion, and excretion by other routes
• A comprehensive discussion of design and optimization of dosage regimens in Chapter 2, including examples of dosing calculations that take into consideration bioavailability, clearance, and distribution
• Appendix II Design Optimization of Dosage Regimens: Pharmacokinetic Data in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition provides a summary of basic pharmacokinetic data for a number of drugs that are in common clinical use, as well as information that is useful in individualizing dosing in a given patient
• Chapter 5 contains a detailed review of membrane transporters, including their role in absorption, distribution, and clearance of xenobiotics, and their role in adverse drug responses
• Table 5-1 Regulation of Transporter Expression by Nuclear Receptors provides detailed information about drug regulation of transporter expression mediated through specific nuclear receptors
• A description in Chapter 5 of the role of efflux transporters in the blood-brain barrier (BBB) and blood-cerebrospinal fluid (CSF) barrier
• Chapter 6 provides a comprehensive discussion of drug metabolism enzymes and illustrations that show many of the key chemical reactions catalyzed by these enzymes
CHARACTERISTICS OF A DRUG THAT PREDICT ITS MOVEMENT AND AVAILABILITY AT ITS SITES OF ACTION
• Molecular size and structural features
• Degree of ionization
• Relative lipid solubility of its ionized and nonionized forms
• Its binding to serum and tissue proteins
LEARNING OBJECTIVES
 Understand key concepts and terms that determine drug pharmacokinetics, including absorption, distribution, metabolism, and excretion.
Understand key concepts and terms that determine drug pharmacokinetics, including absorption, distribution, metabolism, and excretion.
 Understand the mechanisms by which drugs cross membranes and the physicochemical factors that influence this transfer.
Understand the mechanisms by which drugs cross membranes and the physicochemical factors that influence this transfer.
 Be able to predict the ionization state of a drug that is a weak acid or base knowing the drug’s pKa and the pH of the fluid.
Be able to predict the ionization state of a drug that is a weak acid or base knowing the drug’s pKa and the pH of the fluid.
 Describe the role of membrane transporters in drug absorption, distribution, and clearance.
Describe the role of membrane transporters in drug absorption, distribution, and clearance.
 Know the major mechanisms by which drugs are metabolized and excreted from the body.
Know the major mechanisms by which drugs are metabolized and excreted from the body.
 Understand the major pharmacokinetic mechanisms that can result in drug interactions.
Understand the major pharmacokinetic mechanisms that can result in drug interactions.
 Know the pharmacokinetic mechanisms that give rise to interpatient variability in drug response and toxicity.
Know the pharmacokinetic mechanisms that give rise to interpatient variability in drug response and toxicity.
 Understand the clinical pharmacokinetic principles used to calculate dosing required to achieve steady-state drug concentrations in plasma.
Understand the clinical pharmacokinetic principles used to calculate dosing required to achieve steady-state drug concentrations in plasma.
 Understand how route of administration can affect the bioavailability and clearance of drugs.
Understand how route of administration can affect the bioavailability and clearance of drugs.
PASSIVE TRANSPORT VERSUS ACTIVE TRANSPORT ACROSS CELLULAR BARRIERS
• Passive transport is the dominant transport mechanism in the disposition of most drugs (Figure 2-1).
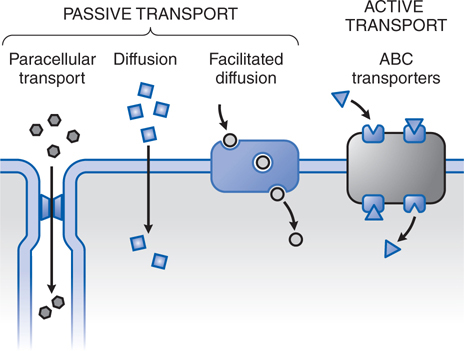
FIGURE 2-1 The variety of ways drugs move across cellular barriers in their passage throughout the body. Transmembrane movement of drug generally is limited to unbound drug; thus drug-protein complexes constitute an inactive reservoir of drug. Unbound drugs cross membranes either by passive processes or by mechanisms involving the active participation of components of the membrane. In passive transfer, the drug molecule usually penetrates by diffusion along a concentration gradient by virtue of its solubility in the lipid bilayer. Such transfer is directly proportional to the magnitude of the concentration gradient across the membrane, to the lipid-water partition coefficient of the drug, and to the membrane surface area exposed to the drug. The greater the partition coefficient, the higher is the concentration of drug in the membrane and the faster is its diffusion. After a steady-state is attained, the concentration of the unbound drug is the same on both sides of the membrane if the drug is a nonelectrolyte. For ionic compounds, the steady-state concentrations depend on the electrochemical gradient for the ion and on differences in pH across the membrane, which will influence the state of ionization of the molecule disparately on either side of the membrane and can effectively trap drug on 1 side of the membrane. Carrier-mediated active transport is characterized by a direct requirement for energy.
▶ In passive diffusion, the drug molecule usually penetrates cellular barriers by diffusion along a concentration gradient by virtue of its solubility in the lipid bilayer.
▶ Paracellular transport through intercellular gaps occurs across capillary endothelium and is important in filtration across the glomerulus in the kidney but is limited in some tissues such as the CNS where capillaries have tight intercellular junctions.
▶ Facilitated diffusion describes a carrier-mediated transport process in which there is no input of energy, and therefore enhanced movement of the involved substance is down a chemical gradient.
• Active transport is characterized by a direct requirement for energy, movement against an electrochemical gradient, saturability, selectivity, and competitive inhibition by cotransported compounds.
In Case 1-1, a gentleman is seeking an allergy medicine at his local pharmacy to relieve his hay fever symptoms. One of the nonprescription drugs he finds contains diphenhydramine, which can cause drowsiness or other CNS effects as discussed in part d of Case 1-1.
a. How is diphenhydramine able to cause CNS effects?
First-generation antihistamines such as diphenhydramine can cross the BBB and cause CNS depression such as somnolence. Diphenhydramine and the other first-generation antihistamines contain a tertiary amino group linked by a 2- or 3-atom chain to 2 aromatic substituents (see Figure 32-3 in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition). The only ionizable group in diphenhydramine and these other agents is the tertiary amino group, which has a pKa ~9.0. Thus, at physiological pH (pH 7.4), the drug is largely unionized (see Figure 2-2) and very lipophilic and can easily diffuse through lipid membranes, including the BBB.
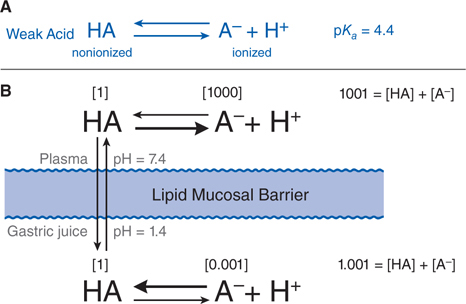
FIGURE 2-2 Influence of pH on the distribution of a weak acid between plasma and gastric juice separated by a lipid barrier. A. The dissociation of a weak acid, pKa = 4.4. B. Dissociation of the weak acid in plasma (pH 7.4) and gastric acid (pH 1.4). The uncharged form, HA, equilibrates across the membrane. Blue numbers in brackets show relative concentrations of HA and A–. In this example, the ratio of nonionized to ionized drug in plasma is 1:1000; in gastric juice, the ratio is 1:0.001, as given in brackets. The total concentration ratio between the plasma and the gastric juice therefore would be 1000:1 if such a system came to a steady-state. For a weak base with a pKa of 4.4 (eg, chlordiazepoxide), the ratio would be reversed, as would the thick horizontal arrows, which indicate the predominant species at each pH. Accordingly, at steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic drug on the more acidic side.
WEAK ELECTROLYTES AND THE INFLUENCE OF PH
• Many drugs are weak acids or bases that are present in solution as both the nonionized and ionized species.
▶ Nonionized molecules are usually more lipid soluble and can diffuse readily across the cell membrane.
▶ Ionized molecules usually are less able to penetrate the lipid membrane because of their low lipid solubility.
• The transmembrane distribution of a weak electrolyte is influenced by its pKa and the pH gradient across the membrane.
• The ratio of nonionized to ionized drug at a given pH is readily calculated from the Henderson-Hasselbalch equation (Equation 2-1), where pKa is the drug’s acid dissociation constant.

• Figure 2-2 illustrates the partitioning of a weak acid (pKa = 4.4) between plasma (pH = 7.4) and gastric juice (pH = 1.4).
• At steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic drug on the more acidic side, a phenomenon known as ion trapping.
• In the kidney tubules where a lipid soluble (uncharged) drug can be reabsorbed by passive diffusion, excretion of the drug can be promoted by altering the pH of the urine to favor the ionized state (A– or BH+).
▶ Alkaline urine favors excretion of weak acids; for example, elevation of urine pH with sodium bicarbonate will increase the excretion of weak acids such as aspirin (pKa ~3.5) and urate (pKa ~5.8).
▶ Acid urine favors excretion of weak bases.
b. Are there other antihistamines that this patient could take that do not cause drowsiness?
The second-generation antihistamines, such as loratadine (see Chapter 21), do not cause drowsiness. These second-generation histamine H1 antagonists are ionized at physiological pH and as a consequence do not readily cross the BBB. For example, the pKa of loratadine is ~4.3, and is thus largely ionized at pH 7.4 and not able to diffuse readily through lipid membranes.
DRUG ABSORPTION, BIOAVAILABILITY, AND ROUTES OF ADMINISTRATION
• Absorption is the movement of a drug from its site of administration into the central compartment (Figure 2-3).
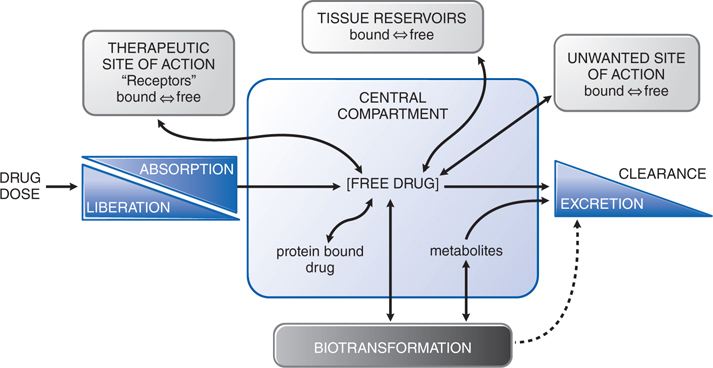
FIGURE 2-3 The interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its sites of action. Possible distribution and binding of metabolites in relation to their potential actions at receptors are not depicted.
• Bioavailability is the fractional extent to which a dose of drug reaches its site of action or a biological fluid from which the drug has access to its site of action.
• A fraction of the administered and absorbed dose of drug will be inactivated or diverted in the intestine and liver before it can reach the general circulation and be distributed to its sites of action.
▶ If the metabolic or excretory capacity of the liver and the intestine for the drug is large, bioavailability will be reduced substantially (first-pass effect).
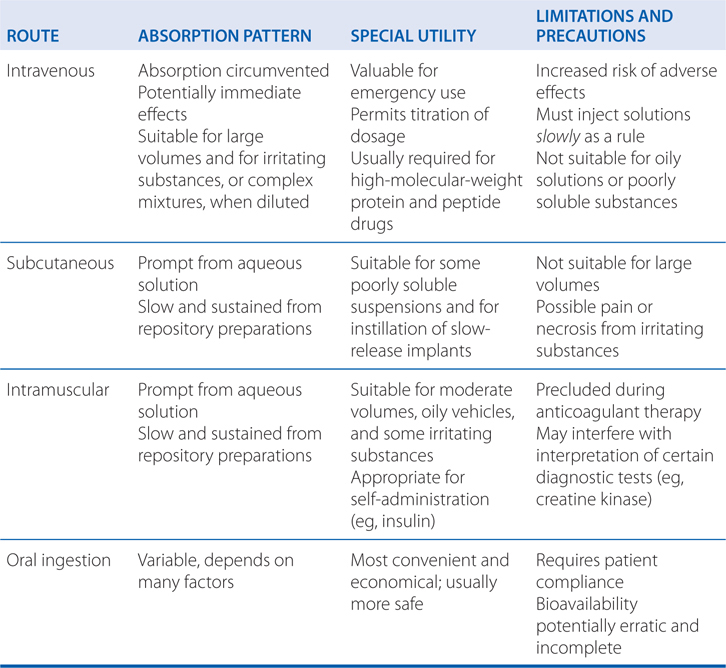
• The route of drug administration can affect the time course of drug effects, extent of absorption, bioavailability, first-pass effect, variability of drug effects, and adverse effects (see Table 2-1).
TABLE 2-1 Some Characteristics of Common Routes of Drug Administration
• Knowledge of drugs that undergo significant metabolism or require active transport across the intestinal and hepatic membranes can be used to predict some pharmacokinetic interactions because drugs may compete for metabolism and transport.
• Distribution refers to the movement of drug into interstitial and intracellular fluids following absorption or systemic administration (see Figure 2-3).
• Distribution reflects a number of physiological factors and the particular physicochemical properties of the individual drug.
• Physiological factors that determine the rate of delivery and potential amount of drug distributed into tissue include:
▶ Cardiac output
▶ Regional blood flow
▶ Capillary permeability
▶ Tissue volume
• During the initial distribution phase, the liver, kidney, brain, and other well-perfused organs receive most of the drug.
• The second distribution phase to muscle, most viscera, skin, and fat is slower and may require minutes to several hours before the concentration of drug in tissue is in equilibrium with that in blood.
▶ The second phase involves a far larger fraction of body mass (eg, muscle) than does the initial phase and generally accounts for most of the extravascularly distributed drug.
• With exceptions such as the brain, diffusion of drug into the interstitial fluid occurs rapidly because of the highly permeable nature of the capillary endothelial membrane.
• Tissue distribution is determined by the partitioning of drug between blood and the particular tissue.
▶ Lipid solubility and transmembrane pH gradients are important determinants of tissue uptake for drugs that are either weak acids or bases; however, ion trapping associated with transmembrane pH gradients is generally not large because the pH difference between tissue and blood (~7.0 vs 7.4) is small.
▶ The most important determinant of blood-tissue partitioning is the relative binding of drug to plasma proteins and tissue macromolecules that limits the concentration of free drug.
c. Besides the CNS, where else in the body might the net charge of a drug be affected by pH?
The partitioning of a weak acid (pKa = 4.4) between plasma (pH = 7.4) and gastric juice (pH = 1.4) is depicted in Figure 2-2. Assume that the gastric mucosal membrane behaves as a simple lipid barrier with a high electrical resistance that is permeable only to the lipid-soluble, nonionized form of the acid. The ratio of nonionized to ionized drug at each pH is readily calculated from the Henderson-Hasselbalch equation (see Equation 2-1). In the example of Figure 2-2, the ratio of nonionized to ionized drug in plasma is 1:1000; in gastric juice, the ratio is 1:0.001, as given in brackets in Figure 2-2. The total concentration ratio between the plasma and the gastric juice therefore would be 1000:1 if such a system came to a steady-state. For a weak base with a pKa of 4.4 (eg, chlordiazepoxide), the ratio would be reversed, as would the thick horizontal arrows in Figure 2-2, which indicate the predominant species at each pH. Accordingly, at steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic drug on the more acidic side.
The effects of net charge are observable elsewhere in the body, such as in the kidney tubules. Urine pH can vary over a ride range, from 4.5 to 8. As urine pH drops (as [H+] increases), weak acids (A–) and weak bases (B) will exist to a greater extent in their protonated forms (HA and BH+); the reverse is true as pH rises, where A– and B will be favored. In the kidney tubules where a lipid-soluble (uncharged) drug can be reabsorbed by passive diffusion, excretion of the drug can be promoted by altering the pH of the urine to favor the ionized state (A– or BH+). Thus, alkaline urine favors excretion of weak acids; acid urine favors excretion of weak bases. Elevation of urine pH (by giving sodium bicarbonate) will promote urinary excretion of weak acids such as aspirin (pKa~3.5) and urate (pKa~5.8). This principle of ion trapping is an important process in drug distribution.
The establishment of concentration gradients of weak electrolytes across membranes with a pH gradient is a physical process and does not require an active electrolyte transport system. All that is necessary is a membrane preferentially permeable to one form of the weak electrolyte and a pH gradient across the membrane. The establishment of the pH gradient, however, is an active process.
A 68-year-old woman has symptoms of angina when she climbs stairs or engages in strenuous activity. She is prescribed nitroglycerin (glyceryl trinitrate, see Chapter 16) to take prophylactically before she engages in any activity that might cause angina symptoms.
a. The patient’s pharmacist instructs her that she is to place the nitroglycerin tablet under her tongue a couple of minutes before strenuous activity to prevent angina. Why is this route of administration used for this drug?
Sublingual administration of nitroglycerin results in rapid onset of action (peak concentrations within 4 minutes of administration) and avoids the first-pass effect. Absorption from the oral mucosa has special significance for certain drugs despite the fact that the surface area available is small. Venous drainage from the mouth is to the superior vena cava, bypassing the portal circulation and thereby protecting the drug from rapid intestinal and hepatic first-pass metabolism. Nitroglycerin is effective when retained sublingually because it is nonionic and has very high lipid solubility. Thus, the drug is absorbed very rapidly. Nitroglycerin also is very potent; absorption of a relatively small amount produces the therapeutic effect (“unloading” of the heart; see Chapter 16). Nitroglycerin that is swallowed undergoes nearly complete first-pass metabolism as the result of enzymatic denitration in the liver. Thus, its bioavailability is only ~0.01 (ie, ~1%) when swallowed.
b. To prevent first-pass effects, what other routes of administration might be effective for this agent?
Because nitroglycerin has very high lipid solubility, it can be absorbed through the skin and administered via controlled-release transdermal patches. This route of administration is effective for patients requiring chronic administration of nitroglycerin to prevent symptoms of angina, but can result in the development of tolerance unless the patch is removed for 8-12 hours each day (see Case 16-1).
PLASMA PROTEIN BINDING OF DRUGS
• Many drugs circulate in the bloodstream bound to plasma proteins.
▶ Albumin is a major carrier for acidic drugs.
▶ α1-Acid glycoprotein binds basic drugs.
▶ Certain drugs may bind to proteins that function as specific hormone carrier proteins, such as the binding of estrogen or testosterone to sex hormone–binding globulin or the binding of thyroid hormone to thyroxin-binding globulin.
• Plasma protein binding is a nonlinear, saturable process.
• Binding of a drug to plasma proteins limits its concentration in tissues and at its site of action because only unbound (free) drug is in equilibrium across membranes (see Figure 2-3).
• Appendix II in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition provides plasma protein binding percentages for a number of commonly used drugs.
• The extent of plasma protein binding also may be affected by disease-related factors.
▶ Changes in protein binding due to disease states and drug–drug interactions are clinically relevant mainly for a small subset of the so-called high-clearance drugs of narrow therapeutic index that are administered intravenously.
• Drug excretion by the kidneys, transport, and metabolism can be limited by binding to plasma protein.
A 59-year-old man who was diagnosed with coronary artery disease undergoes angioplasty and receives a coronary stent. To prevent platelet thrombosis of the stent, he is prescribed a standard dosing regimen of clopidogrel, a drug that inhibits platelet aggregation (see Chapter 19). After several weeks of clopidogrel therapy, his platelet function is measured and it is determined that the dose of clopidogrel is too low to effectively inhibit platelet aggregation.
a. What kind of drug is clopidogrel and how might this explain the poor response to therapy with this agent?
Clopidogrel is a prodrug that requires bioactivation, primarily by CYP2C19. There is wide interindividual variability in the capacity of clopidogrel to inhibit platelet aggregation, and some patients are designated resistant to the antiplatelet effects of the drug. This variability reflects, at least in part, genetic polymorphisms in the CYPs involved in the metabolic activation of clopidogrel, most importantly CYP2C19. Clopidogrel-treated patients with the loss-of-function CYP2C19*2 allele exhibit reduced platelet inhibition compared with those with the wild-type CYP2C19*1 allele and experience a higher rate of cardiovascular events. Even patients with the reduced-function CYP2C19*3, *4, or *5 alleles may derive less benefit from clopidogrel than those with the full-function CYP2C19*1 allele. Although CYP3A4 also contributes to the metabolic activation of clopidogrel, polymorphisms in this enzyme do not appear to influence clopidogrel responsiveness.
TISSUE BINDING OF DRUGS
• Many drugs accumulate in tissues at higher concentrations than those in the extracellular fluids and blood.
• Tissue accumulation may be a result of active transport or, more commonly, binding to cellular constituents such as proteins, phospholipids, or nuclear proteins.
• Tissue binding is generally reversible and can serve as a reservoir that prolongs drug action in that same tissue or at a distant site reached through the circulation.
▶ Tissue binding and accumulation also can produce local toxicity.
• Many lipid-soluble drugs are stored by physical solution in the neutral fat; hence, fat may serve as a reservoir for lipid-soluble drugs.
• Some drugs, divalent metal ion chelating agents, and heavy metals may accumulate in bone by adsorption onto the bone crystal surface and eventual incorporation into the crystal lattice.
• Transporters are membrane proteins that control the influx (uptake) of essential nutrients and ions, and the efflux of cellular waste, environmental toxins, drugs, and other xenobiotics.
• The functions of membrane transporters may be facilitated (equilibrative, not requiring energy) or active (requiring energy); see Figure 2-1.
• The transport of drugs is primarily mediated by 2 major superfamilies, ABC (ATP-binding cassette) and SLC (solute carrier) transporters.
▶ Most ABC proteins are primary active transporters, which rely on ATP hydrolysis to actively pump their substrates across membranes (Table 2-2).
TABLE 2-2 ABC Transporters Involved in Drug Absorption, Distribution, and Excretion Processes
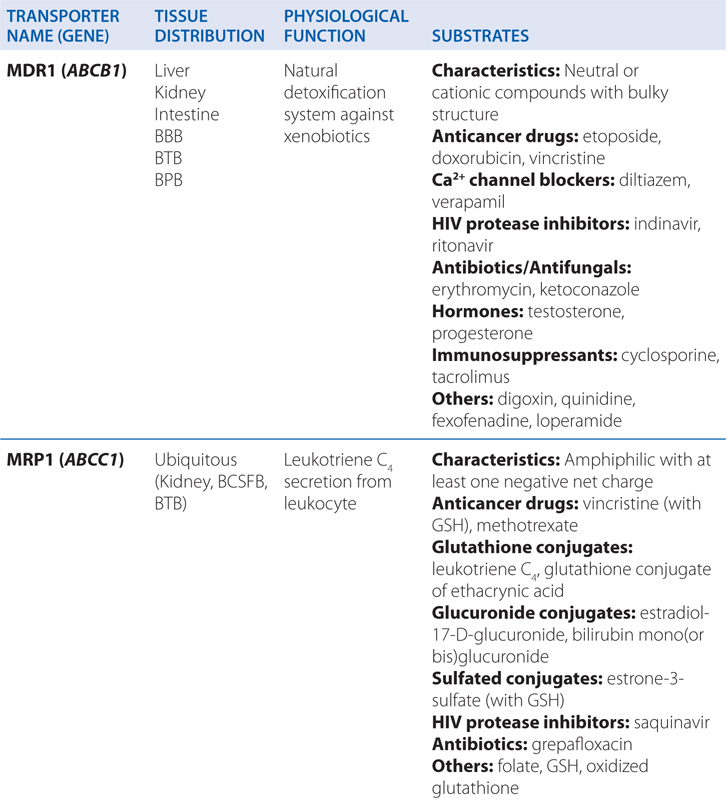


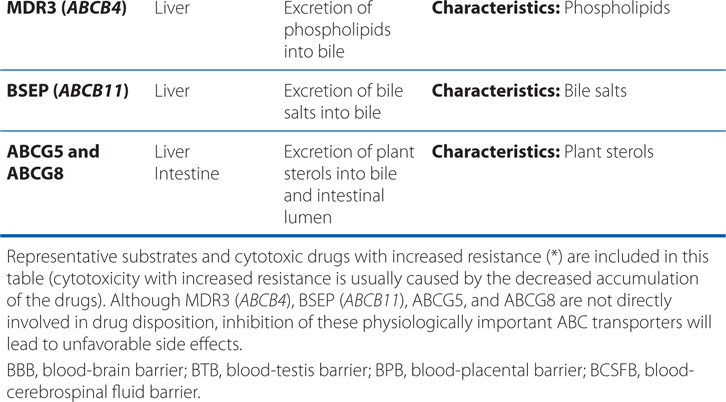
■ There are 49 known genes for ABC protein which include P-glycoprotein (P-gp, encoded by ABCB1, also termed MDR1) and the cystic fibrosis transmembrane regulator (CFTR, encoded by ABCC7).
▶ The SLC superfamily includes genes that encode facilitated transporters and ion-coupled secondary active transporters (Table 2-3).
TABLE 2-3 Families in the Human Solute Carrier Superfamily
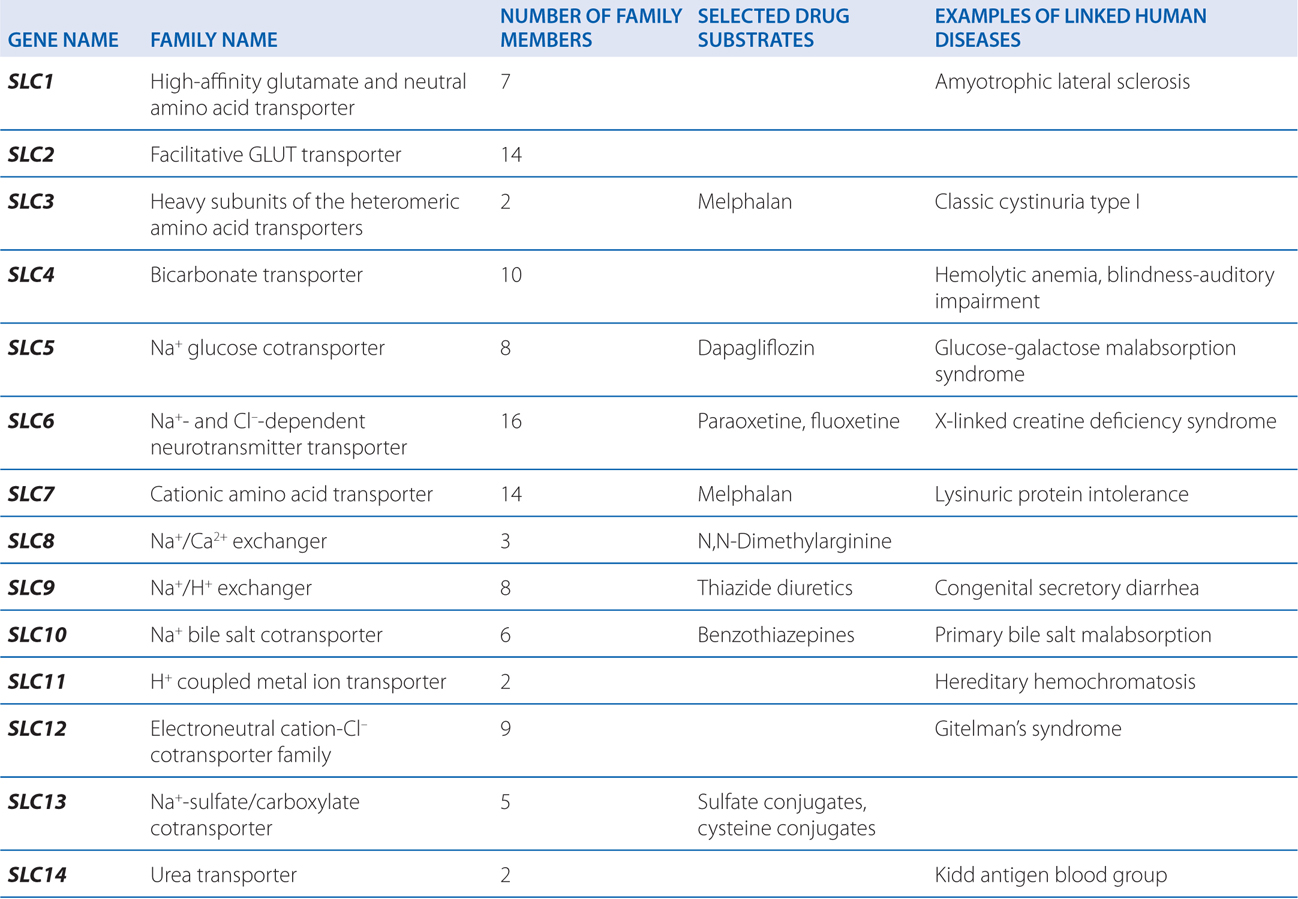
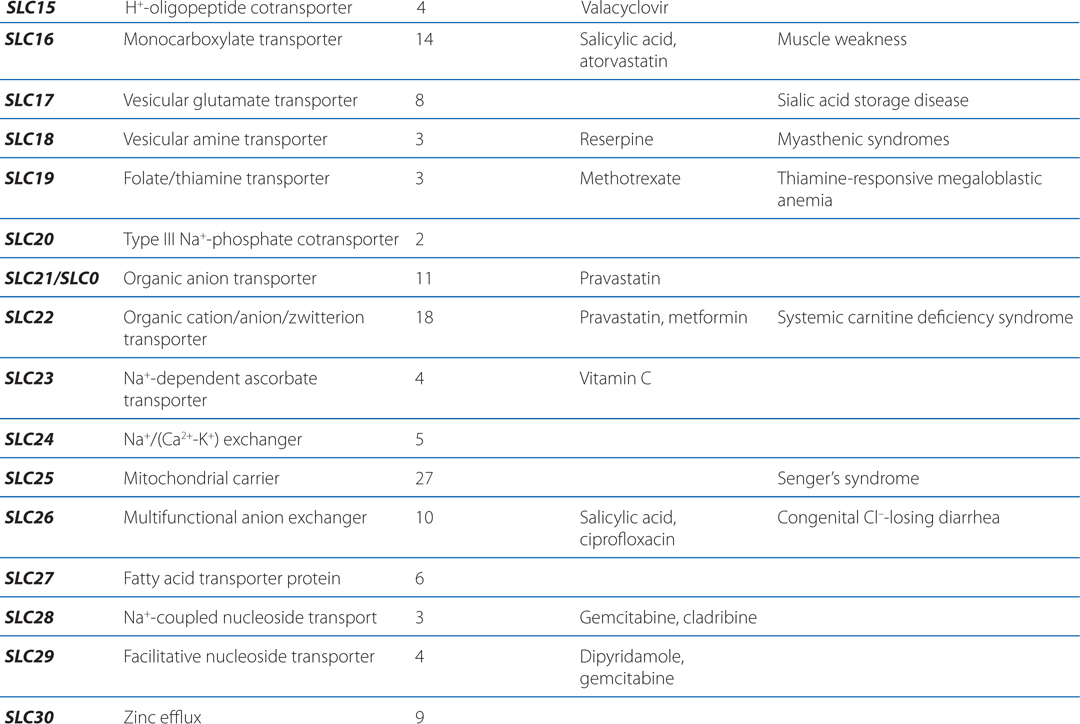
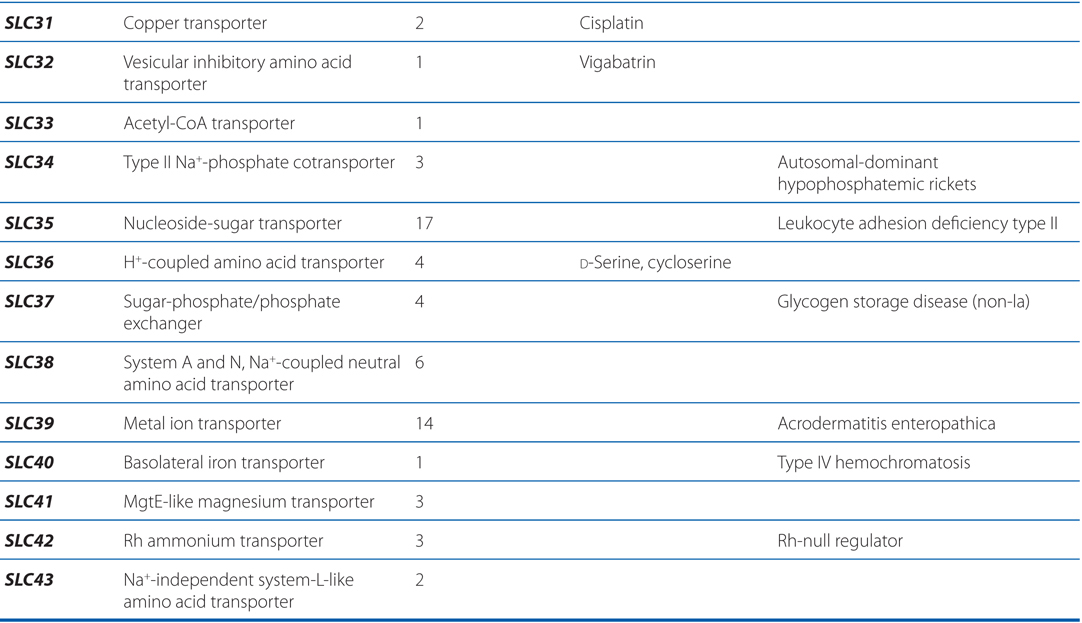
■ Approximately 315 SLC transporters have been identified in the human genome, many of which serve as drug targets or in drug absorption and disposition, including the serotonin (5-HT) and dopamine transporters (SERT, encoded by SLC6A4; DAT, encoded by SLC6A3).
• Uptake and efflux transporters determine the plasma and tissue concentrations of endogenous compounds and xenobiotics, and thereby can influence the systemic or site-specific toxicity of drugs.
• Genetic mutations and polymorphisms in drug membrane transporters can lead to significant variability in individual patient drug disposition and response (for examples, see Table 2-4).
TABLE 2-4 Examples of Genetic Polymorphisms Influencing Drug Response
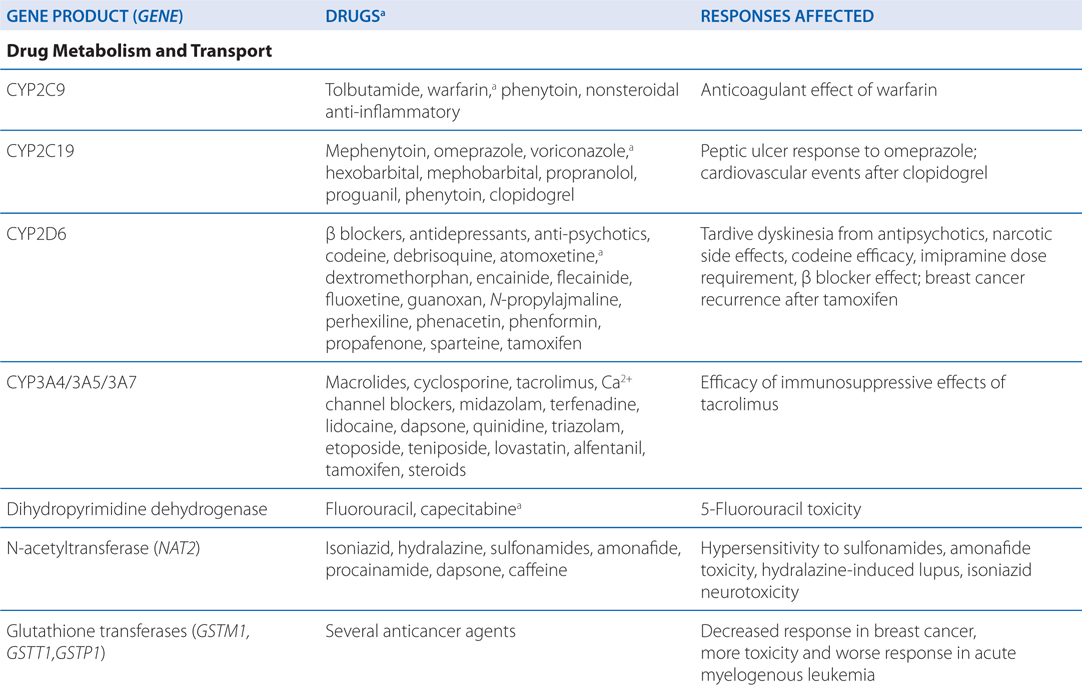
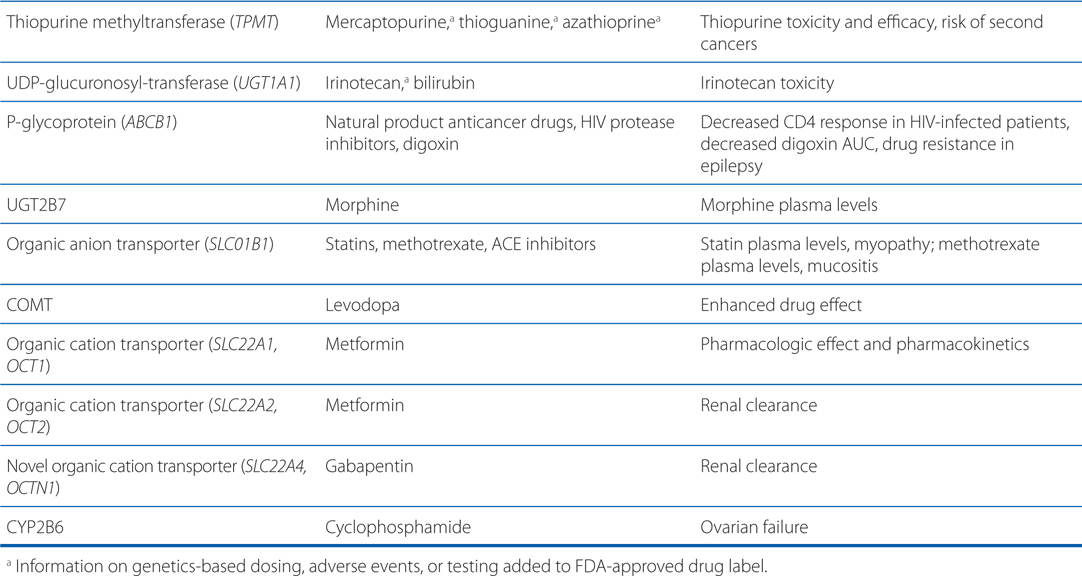
• Distribution of drugs into the CNS from the blood is unique because:
▶ Brain capillary endothelial cells have continuous tight junctions, thus drug penetration into the brain depends on transcellular rather than paracellular transport.
▶ The BBB, which consists of brain capillary endothelial cells and pericapillary glial cells, have unique properties that limit drug transport.
▶ At the choroid plexus, a blood-CSF barrier is present with epithelial cells that are joined by tight junctions, rather than endothelial cells.
• The more lipophilic a drug, the more likely it is to cross the BBB (see Case 2-1).
• Drugs may penetrate into the CNS by specific uptake transporters normally involved in the transport of nutrients and endogenous compounds from blood into the brain and CSF.
• The functional BBB and blood-CSF barrier involve efflux transporters that are capable of removing a large number of chemically diverse drugs from the CNS (see Figure 2-4, and Tables 2-2 and 2-3).
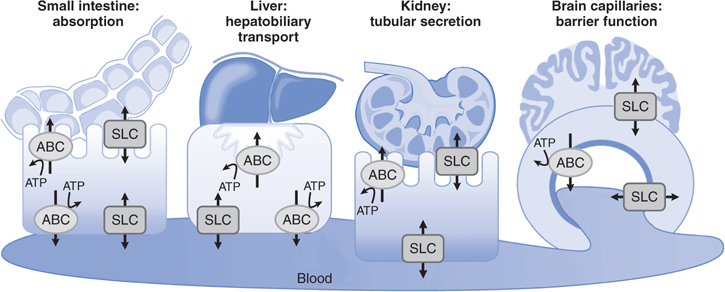
FIGURE 2-4 Transepithelial or transendothelial flux. Transepithelial or transendothelial flux of drugs requires distinct transporters at the 2 surfaces of the epithelial or endothelial barriers. These are depicted diagrammatically for transport across the small intestine (absorption), the kidney and liver (elimination), and the brain capillaries that comprise the BBB. Asymmetrical transport across a monolayer of polarized cells, such as the epithelial and endothelial cells of brain capillaries, is called vectorial transport. Vectorial transport is important in the efficient transfer of solutes across epithelial or endothelial barriers. From the viewpoint of drug absorption and disposition, vectorial transport plays a major role in hepatobiliary and urinary excretion of drugs from the blood to the lumen and in the intestinal absorption of drugs. In addition, efflux of drugs from the brain via brain endothelial cells and brain choroid plexus epithelial cells involves vectorial transport. The ABC transporters mediate only unidirectional efflux, whereas SLC transporters mediate either drug uptake or efflux.
▶ MDR1 (P-gp) and the organic anion–transporting polypeptide (OATP) are 2 of the more notable efflux transporters expressed in the BBB and blood-CSF barrier (BCSFB).
▶ Efflux transporters are expressed in brain capillary endothelial cells and the choroid plexus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


