Anne Zajicek, MD, PharmD, FAAP, and Lori D. Karan, MD, FACP, FASAM
6
Addiction medicine is the application of pharmacologic principles to drugs of abuse. Drugs of abuse are used to produce mood alterations, which requires drug penetration into the brain.
PHARMACOKINETICS
Pharmacokinetics describes the time course of drug concentrations in blood and tissues (e.g., brain). Drug concentrations in blood and other sites are determined by absorption, distribution, metabolism, and elimination.
Absorption
Absorption is the process of drug movement from the site of drug delivery to the site of action. Figure 6.1 illustrates the differences in drug concentrations over time for the various routes of administration. The more rapidly a psychoactive drug is delivered to its site of action in the central nervous system, the greater is its mood-altering and reinforcing effects. The more rapidly achieved and higher the peak concentrations from intravenous and pulmonary (smoking) routes illustrate this point.
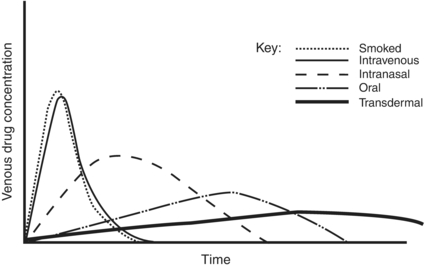
FIGURE 6-1. Venous drug concentrations after different routes of administration.
Bioavailability is defined as the fraction of unchanged drug that reaches the systemic circulation after administration by any route. First-pass metabolism is the metabolism that occurs before a drug reaches the systemic circulation and occurs most extensively for lipid-soluble drugs such as morphine, methylphenidate, and desipramine, and can significantly reduce bioavailability. Morphine, for example, requires nearly twice the dose when administered orally as compared to intravenously.
Distribution
Once absorbed, a drug is distributed to the various organs and tissues of the body.
Most psychoactive drugs enter the brain because they are highly lipid soluble. The blood–brain barrier hinders the ability of non–lipid- soluble drugs to reach the brain tissue by diffusion. For some compounds, specific active transport systems exist. These active transport systems enable glucose, amino acids, amines, purines, nucleosides, and organic acids to gain access to the brain. In contrast, p-glycoprotein is an efflux carrier.
Clearance
Elimination refers to disappearance of the parent and/or active molecule from the bloodstream or body, which can occur by metabolism and/or excretion. Excretion is the process of removing a compound from the body without chemically changing that compound. Metabolic capacity determines drug clearance in most cases.
Most drugs display first-order elimination kinetics: the fraction or percentage of the total amount of drug present in the body removed at any one time is constant and independent of dose. Following administration of a drug with first-order kinetics, concentrations show an exponential decline of drug concentrations. The half-life (t1/2) of a drug is the amount of time it takes for a drug concentration to decrease by half, or conversely achieve half of steady-state concentrations. One half-life represents a 50% change, and 2, 3, 4, and 5 half-lives represent 75%, 87.5%, 93.7%, and 96.8% changes, respectively.
In contrast, for drugs with zero-order elimination kinetics, the amount of drug removed (rather than the fraction of drug removed) at any one time is constant and dependent on dose. The maximal rate of metabolism and/or elimination is generally due to saturation of a key enzyme. Drug dosing becomes difficult in these cases: a small increase in dose can cause a large increase in concentration, in contrast to drugs with first-order clearance where there is proportionality between dose and concentration. Aspirin, phenytoin, and ethanol are examples of drugs with zero-order elimination.
Drug metabolism is the process of chemical modification of drugs and other chemicals by the body, generally into less active and more hydrophilic compounds. These chemical modifications/reactions are generally performed by enzymatic systems, such as the cytochrome P450 enzyme system. Not all metabolites are inactive or nontoxic, and active metabolites need to be considered when assessing a drug’s total activity.
PHARMACOGENOMICS
Pharmacogenomics is the study of the relationship between genetic variations and drug disposition and response.
Genetic variability in drug-metabolizing enzymes can affect drug bioavailability and clearance. Single nucleotide polymorphisms may alter CYP activity. CYP 2D6, for example, which metabolizes codeine to morphine, is the best studied of the drug metabolizing enzymes. Individuals can be genotyped for 2D6 enzyme function (with classification as poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Those with poor metabolizer genotypes generally do not receive adequate analgesia due to the inability to metabolize codeine to the active morphine. Ultrarapid metabolizers, on the other hand, metabolize codeine significantly more rapidly and extensively than others, producing rare but life-threatening morphine intoxication. Breast-feeding infants of mothers who are ultrarapid metabolizers have received morphine overdoses from their mothers who are prescribed codeine for postpartum pain relief.
Drug interactions at the level of the cytochromes and other metabolizing systems are often clinically significant. Methadone is metabolized primarily by CYP 3A4, with contributions from CYP 2B6, 2C19, 3A4, and to a lesser extent CYP 2D6. Inhibitors of CYP 3A4, including erythromycin, diltiazem, ketoconazole, and saquinavir, slow the metabolism of methadone and increase methadone levels. Inducers of CYP 3A4 such as carbamazepine, phenobarbital, efavirenz, and St. John’s wort speed the metabolism of methadone and decrease methadone levels. Awareness of potential interactions, clinical observation, and tailoring medication regimens and dosages are needed to optimize therapy and minimize potential toxicities.
Genetically defined differences in drug metabolism may influence risk of addiction, with relative protection for persons who experience adverse drug reactions at lower drug doses.
PHARMACODYNAMICS
Pharmacodynamics is the study of the dose–response phenomena, which are the biochemical and physiologic effects of drugs on the body, and the body’s homeostatic response. Most drugs act on specific endogenous targets, or receptors, to modulate the rate and extent of the body’s endogenous functions.
Potency, Efficacy, and Dose Response
When drug dose and response are plotted on a logarithmic scale, a sigmoidal curve often results. The maximal efficacy of a drug occurs at Emax, and the concentration of the drug needed to produce 50% of the maximal effect occurs at EC50. Potency denotes the amount of drug needed to produce a given effect; the more potent the drug, the smaller the dose required to achieve maximal effect. Potency is primarily determined by the affinity of the receptor for the drug (Fig. 6.2).
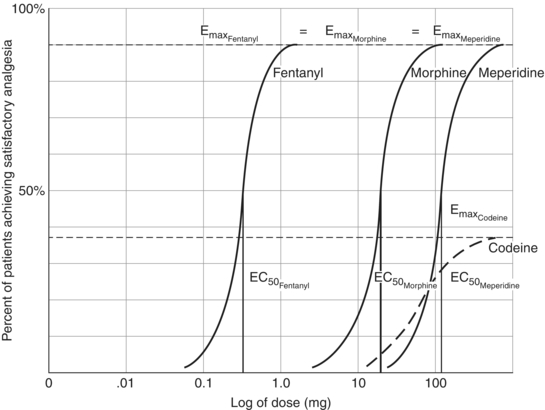
FIGURE 6-2. Dose–response curves of some opioids. Fentanyl has a lower EC50 than does morphine and is more potent than is morphine. Both fentanyl and morphine have a higher Emax than does codeine and are more efficacious than is codeine. Meperidine is less potent than is morphine but more efficacious than is codeine.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


