FIGURE 7-1. Sample polymorphic genes involved in drug pharmacokinetics. Polymorphic genes involved in drug pharmacokinetics are listed next to organs involved in drug absorption, distribution, metabolism, and excretion. VisiScience, Inc. software was used in the creation of the image.
Cytochrome P450 System
While many of the genes encoding CYP enzymes are highly polymorphic, CYP2D6, CYP2C9, and CYP2C19 have genetic variations (polymorphisms), which can describe fairly predictable distributions of drug concentrations, making them clinically relevant for some pharmacogenetic tests. Metabolizer status can be described as extensive (i.e., “normal”), intermediate, poor, or ultra-rapid based on the presence or absence of gene variations. The specific polymorphisms leading to these phenotypes are typically described using star (*) nomenclature as defined by the Cytochrome P450 Nomenclature Committee (http://www.cypalleles.ki.se), whereby the *1/*1 genotype is considered the common or normal, fully-functioning form of the gene. This genotype–phenotype relationship could help identify poor metabolizers likely to experience side effects (or therapeutic failure in the case of prodrugs requiring activation) to usual doses of CYP450-metabolized drugs or ultra-rapid metabolizers more prone to therapeutic failure (or toxicity in the case of prodrugs). The doses of these individuals with altered metabolism could then be increased or decreased as appropriate or alternative drugs chosen. This ability of genotype to predict drug metabolizing phenotype may be especially important for drugs with narrow therapeutic indices and less important for wide therapeutic index drugs.11
CYP2D6
The CYP2D6 gene contains over 80 alleles, which can lead to normal-functioning, reduced- or nonfunctional protein, or even multiple copies of the gene. The most common nonfunctional alleles are *3, *4, *5, and *6. One of the most reproducible associations between CYP2D6 genotype and a drug response occurs with codeine. Codeine is a prodrug requiring metabolism by CYP2D6 into its active form, morphine, for its analgesic effect. Therefore, individuals who are CYP2D6 poor metabolizers are at risk of therapeutic failure and those who are ultra-rapid metabolizers are at risk of toxicity. Alternative analgesic therapy is recommended in both of these groups of patients. Guidelines have been published with recommendations for CYP2D6 genetic testing interpretation and suggested clinical action for the test results.12
CYP2C19
The CYP2C19 gene contains over 25 alleles leading to normal-, reduced-, non-, or over-functioning protein. The most common nonfunctional alleles are *2 and *3, which account for 85% of reduced function alleles in Caucasians and Africans and 99% of reduced function alleles in Asians. The other reduced or nonfunctional alleles, *4 to *8, are less common. The *17 allele is a gain-of-function allele and has a frequency of 3% to 20% depending on ethnicity.
One of the most clinically actionable associations between CYP2C19 polymorphisms and a drug response is with clopidogrel. Clopidogrel is a prodrug requiring activation by two CYP450-dependent steps, both of which involve CYP2C19. Individuals carrying reduced function CYP2C19 alleles have been shown to have lower active metabolite concentrations, reduced inhibition of platelet aggregation, and increased risk of adverse cardiovascular outcomes when treated with clopidogrel at standard doses compared to those without reduced function alleles.13-17 Based on these data, the FDA updated the clopidogrel label to indicate that individuals with two reduced function CYP2C19 alleles should receive alternative treatment or treatment strategies. Guidelines have been published with treatment recommendations based on CYP2C19 genotype.18
CYP2C9
CYP2C9 contains over 30 alleles that lead to decreased or nonfunctional protein. The most common variants in CYP2C9 are the *2 and *3 alleles. The *2 allele has a frequency of approximately 13% in whites, 0% in Asians, and 3% in blacks. The *3 allele has a frequency of approximately 7% in whites, 4% in Asians, and 2% in blacks. The most well-documented association with CYP2C9 is with warfarin dose requirements (discussed below).
Other CYP450s
CYP3A4 contains over 30 reported polymorphisms, but these variations result in a unimodal distribution of drug clearance, lending themselves less well to use in the clinic setting. None of the discovered CYP3A4 variants, with the exception of one rare variant—*20 (which has a frequency of less than 0.6% in Europeans)—result in nonfunctional protein.19 This unimodal distribution likely results from the small contribution each individual polymorphism in the gene makes to phenotypic variation and the fact that environmental factors may play a bigger role in CYP3A4 activity than with other enzymes. CYP3A5 has proved to have more predictable associations between polymorphisms and expression of CYP3A5 enzyme. Roughly 10% to 20% of whites, 85% of blacks, 60% of Hispanics, and 50% of east Asians have genetic variants in CYP3A5 that cause them to express CYP3A5 hepatically and intestinally.20 Consequently, this proportion of individuals may require dose modifications of CYP3A5-metabolized drugs, and in fact CYP3A5 genetic variants have been implicated in variable drug responses for many drugs.
Thiopurine Methyltransferase
Thiopurine methyltransferase (TPMT) is the enzyme responsible for the conversion of azathioprine and 6-mercaptopurine into inactive metabolites (Figure 7-2). Genetic variants in the TPMT gene can result in deficient or absent TPMT activity leading to severe hematological adverse effects with azathioprine or 6-mercaptopurine (6-MP) treatment. The wild-type (common) allele in TPMT is designated TPMT*1. The most common variants in TPMT are referred to as TPMT*2, *3A, and *3C and are derived based on the presence or absence of any of three single nucleotide variants in the gene (G238→C; G460→A; and A719→G). Approximately one in 300 individuals possess two copies of these variant alleles and therefore lack TPMT activity. These individuals require dose reductions of thioguanines like 6-MP on the magnitude of 90% to avoid hematological toxicity. Individuals with one copy of a variant allele make up about 10% of the Caucasian population and require dose reductions of approximately 50%.
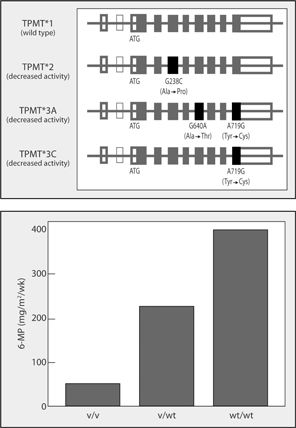
FIGURE 7-2. TPMT polymorphisms and 6-mercaptopurine dose requirements. (A; top) The TPMT gene structure is shown with exons indicated by boxes and introns as the horizontal line. The thin line box indicates exon 2, which is rarely expressed in the general population. Four major TPMT alleles are shown. The wild-type allele encodes a fully active enzyme, whereas the variant alleles TPMT*2, TPMT*3A, and TPMT*3C, which together account for >95% of intermediate- and low-activity variants, encode proteins with decreased enzymatic activity. (B; bottom) Variable 6-MP dosage requirements for individuals with homozygous variant (v/v), heterozygous (v/wt), and homozygous wild-type (wt/wt) alleles. (Source: Reprinted from reference 70.)
Azathioprine and 6-MP are used in the treatment of childhood acute lymphoblastic leukemia (ALL), rheumatoid arthritis, prevention of renal allograft rejection, and in the management of autoimmune disorders. In 2004, the FDA added language to the package insert of 6-MP indicating that TPMT genotyping or phenotyping should be considered prior to treatment.21 Many major academic cancer hospitals routinely perform TPMT activity testing prior to 6-MP dosing for this indication and practice guidelines have been published.22 Individuals with two deficient TPMT alleles (or deficient activity) require 10-fold starting dose reductions in order to avoid severe myelosuppression (recommendation classification strong).22 Individuals with one deficient allele (or intermediate activity) have more variable dose requirements, with 30% to 60% of heterozygotes being unable to tolerate full doses. Starting dose reductions of 30% to 50% are recommended in heterozygotes (recommendation classification moderate).22 Thiopurine methyltransferase activity can be determined either by enzymatic testing of red blood cell lysate or by genotyping. A study addressed the prevalence of TPMT enzyme and genotype testing across a national survey of dermatologists, gastroenterologist, and rheumatologists in England.23 Overall, TPMT enzyme testing was reportedly used by 67% of respondents. This testing was most frequently used by dermatologists (94%), followed by gastroenterologists (60%), and rheumatologists (47%). Genotype testing was not routinely available to practitioners participating in this survey and, hence, was only used by 5% of respondents.
It is important to note that the measurement of TPMT enzyme activity can be impacted by concurrent drugs such as salicylates and by recent blood transfusions. There has been some debate regarding whether genotyping or measurement of enzyme activity is superior.24 Genotyping may be more useful at the start of therapy to minimize the likelihood of life-threatening toxicity, in the setting of blood transfusion (as phenotyping is inaccurate), and in bowel disease or rheumatoid arthritis where, unlike in leukemia, serial blood draws are not routinely performed. Phenotyping may be more useful after a disease flare (as a compliance measure), after several months of therapy (TPMT induction effect has been described), or at the time of an adverse event.
Dihydropyrimidine Dehydrogenase
Dihydropyrimidine dehydrogenase (DPD) metabolizes 5-fluorouracil, an agent commonly used in the treatment of solid organ tumors. In the mid-1980s, it was recognized that deficiencies in DPD were heritable and associated with severe 5-fluorouracil toxicity.25,26 Many polymorphisms in the gene encoding DPD, DPYD, have been identified, although very few of them have any effect on enzyme activity. An intronic polymorphism, IVS14 +1 G>A (DPYD*2A) has been the most widely studied and has been found in approximately half of cases of DPD deficiency. Other common polymorphisms, which have been assessed and found in cases of DPD deficiency, are 2846 A>T and 1679 T>G.
As with TPMT testing, DPD deficiency can be tested for genetically or with enzymatic testing. The analytical performance of using genetic testing to predict toxicity has ranged from 6.3% to 83% for sensitivity, 82% to 100% for specificity (although specificity was not determinable in most studies evaluated), 62% to 100% for positive predictive value, and 86% to 94% for negative predictive value (again, not determinable for most studies).27 Fewer studies have evaluated the test performance characteristics of enzymatic testing, but two that did had a sensitivity of 60%.27
Clinical Significance
When deciding whether drug metabolism polymorphisms might be clinically significant for particular drugs, three main factors should be considered. First, is the drug metabolism enzyme of interest an important route of elimination for the drug in question? If not, even functional polymorphisms in this gene are not going to have a great impact on the pharmacokinetics of the drug. Second, does the medication of interest have a narrow therapeutic index or steep exposure/response curve? If not, changes in plasma concentrations may not be great enough to influence the dose-response relationship. Last, are other therapeutic alternatives available to the medication in question? If so, these alternatives may have other routes of metabolism that are not polymorphic, and the variability in pharmacokinetics could be avoided altogether.
DRUG-TARGET-RELATED MOLECULAR TESTS
While pharmacokinetics is concerned with ADME, pharmacodynamics is concerned with drug effects on target molecules, tissues, and physiological processes. There are abundant examples of pharmacogenetic studies in the literature, where the candidate gene of interest was one related to drug target physiology (vis-à-vis drug metabolism).5 Furthermore, there are illustrative examples in cardiology and oncology where consideration of genetic variation may improve drug therapy. In oncology, in addition to germline (inherited) variations, tumor cells can also exhibit acquired or noninherited (somatic) variations only present in the tumor tissue. This adds a layer of complexity to molecular testing, involving the acquisition and processing of tumor tissue with adequate quality and quantity to accommodate the test of interest. Moreover, laboratories performing specialized testing should have demonstrated proficiency in the specific technology being used. This section deals with five such drugs: the anticoagulant warfarin, the breast cancer drug trastuzumab, the lung and pancreatic cancer drug erlotinib, the lung cancer drug crizotinib, and the melanoma drug vemurafenib.
Warfarin is the most commonly prescribed anticoagulant for the treatment and prevention of thrombosis. Despite its widespread use, warfarin has a narrow therapeutic range (as measured by the international normalized ratio [INR]) below which thrombosis risk is increased and above which bleeding risk is increased. While patient-specific factors such as age, sex, race, and diet partially explain variability in warfarin response, these factors don’t reliably predict the likelihood for efficacy or bleeding risk. As such, investigators have studied the role of genetic variants in an enzyme responsible for warfarin’s metabolism (CYP2C9) and target gene (VKORC1) on warfarin responses. These studies have investigated the impact of genetic and nongenetic factors on endpoints related to INR, bleeding, and clinical efficacy.
CYP2C9 is the major metabolic route for the more potent warfarin enantiomer. The CYP2C9*1 allele is associated with full metabolic capacity, while the well-studied *2 and *3 alleles are associated with decreased activity, diminished warfarin clearance, and lower warfarin dose requirements.28-33 By extension, these variant carriers exhibit longer than normal time to achieve target INR and are at increased risk for bleeding.34 More recently, pharmacogenetic studies have also included analysis of VKORC1 polymorphisms. Taken in sum, CYP2C9 and VKORC1 polymorphisms when considered with clinical correlates of warfarin dose explain approximately 50% of the variability in warfarin dose requirements.35,36 Dosing algorithms including CYP2C9, VKORC1, and clinical information are continually being developed and tested, and the FDA updated the warfarin label with estimated doses by genotype.37-39 The Couma-Gen study demonstrated the feasibility of performing prospective comparisons of pharmacogenetic-guided versus traditional dosing of warfarin, and the National Institutes of Health has a large prospective analysis of warfarin pharmacogenetics that is underway.40 Guidelines have also been published with recommendations for clinical interpretation of warfarin pharmacogenetics data.39
Trastuzumab
Trastuzumab is a monoclonal antibody against the human epidermal growth factor receptor 2 (HER2), encoded by the ERBB2 gene and represents a modality for the treatment of breast cancer. It was noted in the early drug development process that trastuzumab was only effective in a subset of patients. It was subsequently elucidated that trastuzumab is only effective in breast cancers that overexpress the HER2 protein, representing approximately 25% of breast cancer tumors. Consequently, treatment with trastuzumab is predicated on this molecular diagnostic and accurate classification of HER2 tumor status is necessary for optimum treatment of HER2-positive tumors. While not a test for genetic polymorphisms per se, HER2 testing is performed by methods including immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) to determine the HER2 overexpression and gene amplification, respectively.41 Both IHC and FISH assays have been approved by the FDA for HER2 testing.42
Clinically, tumor biopsies are obtained from individuals with breast cancer in which trastuzumab may be an option. Immunohistochemistry is often used as initial screening for HER2/neu overexpression because of its more routine availability in clinical laboratories and lower cost than FISH. Guidelines regarding HER2 testing have recently been updated. 42,43 In one algorithm, a semiquantitative IHC test is performed on the tissue sample to determine the extent of HER2 overexpression. Samples with scores of 0/1+ are considered negative; those with scores of 2+ are borderline/equivocal; 3+ tissues are considered positive.42,44 In cases of borderline specimens, FISH is recommended for subsequent evaluation with a FISH ratio greater than 2.0 generally considered a positive test.
The test performance characteristics of IHC and FISH have been well described. For example, in analysis of approximately 3,000 breast cancer specimens, the positive predictive value of IHC (3+) was 92%, and the negative predictive value (0 or 1+) was 97%.45 These molecular tests are widely available and used clinically in the setting of breast cancer treatment with trastuzumab. Furthermore, the techniques are not limited to breast tumors and are likely to be extended to other disease states and drug therapies in which gene product expression is a determinant of clinical responsiveness.
More recently, trastuzumab has been approved in combination with chemotherapy as an option for the treatment of patients with HER2-overexpressing metastatic gastric cancer, or gastroesophageal junction (GEJ) adenocarcinoma. Due to differences in tumor histopathology (breast or gastric/GEJ adenocarcinoma), tests developed for the specific tumor type to assess HER2 protein overexpression and HER2 gene amplification should be used.46
Erlotinib
Erlotinib is an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). Much like HER2 discussed above, EGFR aberrant signaling is associated with development and prognosis of certain cancers, and EGFR inhibition with drugs such as erlotinib results in blockage of important processes in the pathogenesis of these cancers. Erlotinib is currently used for the treatment of non-small cell lung cancer (NSCLC) and pancreatic cancer.
Increased sensitivity to EGFR-TKIs in NSCLC has been linked to the presence of EGFR activating mutations in the tumor (mostly in exons 18 to 21 of the EGFR gene). These mutations are most common in women, Asian individuals, never-smokers, and patients with adenocarcinoma histology.47 In a 2004 landmark study, Lynch and colleagues identified mutations in the EGFR gene in tumors of patients with NSCLC who were responsive to gefitinib, another EGFR-TKI.48 Sensitivity and specificity were 89% and 100%, respectively; positive and negative predictive values were 100% and 88%. Since this publication, several clinical trials have prospectively tested the impact of somatic EGFR-activating mutations on clinical response to TKIs (e.g., gefitinib and erlotinib) among lung cancer patients. Results from these studies have underscored the importance of EGFR mutation testing in guiding therapy choices for advanced NSCLC.49,50 Approximately 20% of lung adenocarcinomas harbor an EGFR activation mutation (this frequency can be higher in East Asians), and several methods are being used to detect these mutations in tumor tissues with different performance characteristics.51
Crizotinib
Crizotinib is a multitargeted receptor tyrosine kinase inhibitor of anaplastic lymphoma kinase (ALK) and potentially other kinases. Different lung cancer subsets can be molecularly defined by the presence of specific “driver” mutations (e.g., EGFR-activating mutations), which are key in tumor pathogenesis. These driver molecular alterations are being targeted for therapy, and the prospective genotyping of lung cancers is becoming a new standard of care.52 One of these unique molecular subsets is characterized by rearrangements involving the ALK gene. In NSCLC, the EML4-ALK is the most commonly reported ALK rearrangement. The resulting fusion gene encodes a cytoplasmic chimeric protein with constitutive kinase activity and tumor promoting potential. EML4-ALK occurs in approximately 5% of unselected NSCLC, and is more common in never- or light-smokers, in young adult patients, and in patients with adenocarcinomas. Multiple distinct EML4-ALK chimeric variants, as well as other fusion partners have been reported.53
Crizotinib was developed along with a companion diagnostic and is indicated for locally advanced or metastatic NSCLC with ALK rearrangements, as detected by an FDA-approved test. Therefore, the detection of ALK-positive NSCLC is necessary for selection of patients for treatment with crizotinib. The companion diagnostic test currently available is a FISH Break Apart assay. This assay employs one probe 5’ of the ALK locus and one probe within the ALK gene. When the probe set is hybridized against normal nuclei, it generates a merged (green–red fluorescent) signal that can be visualized microscopically. However, the signal is split when the probe set is hybridized against nuclei with a rearrangement involving the 5’ portion of the ALK locus. To be considered positive for ALK rearrangements and eligible for treatment with crizotinib, at least 15% of the tumor cells analyzed have to harbor either the break-apart signals or have a single 3’ ALK (red) signal, 50 cells or more have to be counted, and the separation between the 5’and 3’ ALK probes has to be at least two signal diameter.54,55 Other tests are being developed as alternatives to FISH, including IHC and reverse transcription polymerase chain reaction (RT-PCR), which has the potential of identifying fusion variants missed by FISH.56,57
Vemurafenib
Vemurafenib is a BRAF serine-threonine kinase inhibitor indicated for patients with advanced melanoma harboring a somatic V600E mutation in the BRAF gene. Mutated BRAF proteins often have elevated kinase activity leading to aberrant activation of survival and anti-apoptotic signaling pathways in the tumor cells.58 As with crizotinib, vemurafenib was also developed along with a companion diagnostic.59 The assay is a PCR-based system that detects BRAF V600E mutations in paraffin-embedded, formalin-fixed tumor samples. Approximately 40% to 60% of cutaneous melanomas are positive for BRAF V600 mutations. Among these, the V600E mutations constitute 80% to 90% of reported V600 BRAF mutations, but other mutations such as V600K and V600D can also occur in this disease setting. During vemurafenib’s phase III pivotal trial, 10 BRAF V600 positive patients (as identified by the Cobas test) were subsequently found to harbor the V600K mutation using DNA sequencing. Four of these patients still had partial responses to the drug.59 These results suggest that melanoma patients with V600 mutations other than V600E such as V600K may still be sensitive to vemurafenib and highlight the importance of understanding the performance of tests used to determine eligibility to therapy. Vemurafenib has not been studied in patients with wild-type BRAF melanoma, and is not recommended for this population.
Resistance to Targeted Therapy
Only a percentage of patients respond to targeted therapies. Moreover, responders often develop resistance. Elucidating underlying mechanisms of primary or acquired resistance at a molecular level is an intense area of research in oncology. For example, somatic point mutations in the KRAS gene, most commonly found in codons 12 and 13, have been strongly associated with primary resistance to the anti-EGFR monoclonal antibodies panitumumab and cetuximab in colorectal cancer. Therefore, these antibodies are not indicated for colorectal cancer patients with tumors positive for these mutations. Of note, different KRAS mutations may not predict in the same extent resistance to anti-EGFR antibodies.60 Point mutations, gene amplifications, changes in protein expression and activation of alternate pathways are among the mechanisms implicated in resistance.61 Molecular assays are being developed to identify these alterations and evaluate their clinical significance.
IMMUNE-RELATED MOLECULAR TESTS
Variants in immune-related genes are increasingly being associated with drug-induced adverse events. One of the most noted examples is that of abacavir. Immune-mediated hypersensitivity reactions occur in 5% to 8% of abacavir-treated patients, usually within the first 6 weeks of treatment. Retrospective case-control studies identified the major histocompatibility complex (MHC) Class I region as being associated with this hypersensitivity reaction.63 Subsequent, prospective randomized-controlled trials demonstrated that screening for the HLA-B*5701 allele eliminated immunologically-confirmed hypersensitivity reaction with a negative predictive value of 100% and a positive predictive value of 47.9%.64 Current HIV treatment guidelines recommend screening for HLA-B*5701 prior to the initiation of an abacavir-containing treatment regimen.65
Clopidogrel Pharmacogenetics
CHARLES B. IS A 59-YEAR-OLD African-American male who presents to the emergency department with chest pain. His cardiac enzymes and electrocardiogram are consistent with an acute coronary syndrome (ACS) and a percutaneous coronary intervention (PCI) with stent implantation is immediately planned. A decision is made to start Charles B. on clopidogrel per the hospital’s standard ACS protocol.
PMH
Hypertension × 20 years
Hyperlipidemia × 10 years
Social history
Smoked 1.5 packs per day × 38 years; quit 10 years ago
Medications
HCTZ 25 mg daily
Verapamil SR 240 mg daily
Atorvastatin 40 mg daily
Weight: 77 kg
Height: 68”
BP: 138/88
HR: 90
Question: Variations in which candidate genes could be expected to impact clopidogrel effectiveness?
Answer: Clopidogrel is a prodrug that needs to be biotransformed to an active metabolite to exert its antiplatelet activity. The metabolism of clopidogrel to its active metabolite is a two-step process that involves the following cytochrome P450s: CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP3A4/5. Variations in any of these genes could conceivably be associated with variable formation of the active metabolite with consequent variability in antiplatelet effects and clinical efficacy. In fact, several of these genes have been studied from a pharmacogenetic perspective, with CYP2C19 being the most widely studied and consistently associated. In addition to drug metabolism enzymes, the p-glycoprotein transporter, encoded by the ABCB1 gene has also been found in some studies to influence clopidogrel response and the paraoxonase I, encoded by PON1, has been identified in one study but not replicated in others. From a pharmacodynamic perspective, the P2Y12 platelet receptor (encoded by P2RY12) is the target of clopidogrel activity and has been associated with clopidogrel efficacy in some studies, although with no robust associations that currently influence treatment recommendations.
Charles B. had been previously seen in a progressive cardiovascular clinic for the management of his hypertension and dyslipidemia. As part of routine practice, preemptive genotyping for “VIPs” (Very Important Pharmacogenes) was performed several months ago. Charles B.’s CYP2C19 genotype status according to his electronic medical record is as follows: CYP2C19 *2/*2 (homozygous for loss-of-function allele).
Question: How would this genotype be expected to impact clopidogrel effectiveness in Charles B.?
Answer: CYP2C19 is involved in both steps of clopidogrel’s two-step transformation to its active metabolite. CYP2C19 is polymorphic with common alleles including *1 (normal activity), *2 and *3 (loss of activity), and *17 (increased activity). Patients are assigned the following likely drug metabolism phenotypes based on their genotypes: ultra-rapid metabolizer (UM): *1/*17, *17/*17; extensive metabolizer (EM): *1/*1; intermediate metabolizer (IM): *1/*2, *1/*3, *2/*17, *3/*17; poor metabolizer (PM): *2/*2, *2/*3, *3/*3. Charles B. is a PM, and PMs are prevalent in about 2% to 15% of the population and have the lowest levels of clopidogrel active metabolite, antiplatelet activity, and worse cardiovascular outcomes compared with EMs. Because of a growing body of evidence suggesting that PMs are at significant risk for not experiencing a treatment benefit when given clopidogrel, the FDA updated the clopidogrel label with a boxed warning in 2010. Specifically, the label notes that “[PMs] treated with [clopidogrel] at recommended doses exhibit higher cardiovascular event rates following ACS or PCI than patients with normal CYP2C19 function” and that one should “consider alternative treatment or treatment strategies in patients identified as CYP2C19 [PMs].”
Question: What possible courses of action are there for people with Charles B.’s genotype in his clinical context?
Answer: The FDA recommends alternative treatments or treatment strategies for known CYP2C19 PMs in the ACS or PCI contexts. Alternative treatments may include antiplatelet agents that are not susceptible to polymorphic metabolism in a clinically significant way (e.g., prasugrel or ticagrelor). Alternative treatment strategies might include increased clopidogrel doses beyond the standard 75 mg daily dose. Pharmacodynamic studies suggest that increased clopidogrel doses of 225 mg per day in IMs achieve platelet inhibition comparable to the standard 75mg dose in EMs, but that increased doses up to 300 mg per day cannot achieve comparable platelet inhibition in PMs.62 Currently, the CPIC recommends PMs and IMs receive prasugrel or alternative therapy if no contraindications exist (classification of recommendation: strong for PMs, moderate for IMs). The guidelines recommend UMs and EMs receive clopidogrel at the standard label-recommended doses.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


