For many drugs, the action of a cytochrome P-450 is to begin the process of detoxification through a series of reactions that render the drug less active and easier to excrete. Some drugs, however, are themselves inactive prodrugs whose conversion into an active metabolite by a cytochrome P-450 is required for the drug to have any therapeutic effect.
Many of the CYP genes important for drug metabolism (including CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) are highly polymorphic, with alleles that result in absent, decreased, or increased enzyme activity, thereby affecting the rate at which many drugs are metabolized, with real functional consequences for how individuals respond to drug therapy (see Table 18-3). As one example, CYP2D6, the primary cytochrome in the metabolism of more than 70 different drugs, has dozens of reduced, absent, or increased activity alleles, leading to normal, poor, or ultrafast metabolism (see Table on metabolizer phenotypes later). Missense mutations decrease the activity of this cytochrome; alleles with no activity are caused by splicing or frameshift mutations. In contrast, the CYP2D6*1XN allele is actually a series of copy number variation alleles in which the CYP2D gene is present in three, four, or more copies on one chromosome. Predictably, these copy number polymorphisms produce high levels of the enzyme. There are dozens more alleles that do not affect the function of the protein and are therefore considered to be wild-type. Various combinations of these four classes of alleles produce quantitative differences in metabolizing activity, resulting in three main phenotypes: normal (also called “extensive”) metabolizers, poor metabolizers, and ultrafast metabolizers (Fig. 18-3).
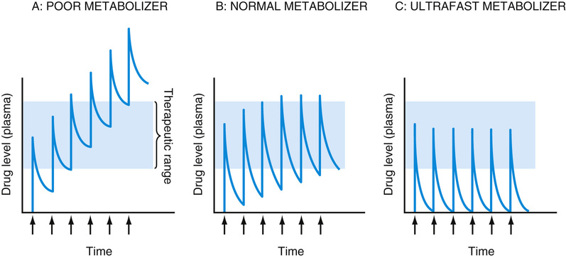
Depending on whether a drug is itself an active compound or is a prodrug that requires activation by a cytochrome P-450 enzyme to have its pharmacological effect, poor metabolizers may either accumulate toxic levels of the drug or fail to have therapeutic efficacy because of poor activation of a prodrug. In contrast, ultrafast metabolizers are at risk for being undertreated by a drug with doses inadequate to maintain blood levels in the therapeutic range, or they may suffer overdose due to too rapid conversion of a prodrug to its active metabolite. For example, codeine is a weak narcotic drug that exerts most of its analgesic effect on conversion to morphine, a bioactive metabolite with a 10-fold higher potency. This conversion is carried out by the CYP2D6 enzyme. Poor metabolizers, quite common in some populations, carrying loss-of-function alleles at the CYP2D6 locus fail to convert codeine to morphine and thereby receive little therapeutic benefit; in contrast, ultrafast metabolizers can become rapidly intoxicated with low doses of codeine. A number of children have died from codeine overdoses due to having an ultrafast metabolizer phenotype.
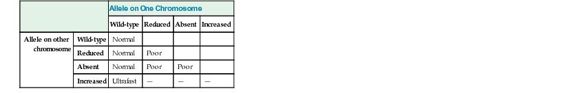
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


