Chapter 16 Pharmaceutical development
Preformulation studies
The main components of preformulation studies are:
• Development of a suitable spectroscopic assay method for determining concentration and purity
• Determination of solubility and dissolution rates of parent compound and salts in water and other solvents
• Chemical stability of parent compound and salts in solution and solid state
• Determination of pKa and pH dependence of solubility and chemical stability
• Determination of lipophilicity (i.e. oil:water partition coefficient, expressed as Kd)
• Determination of particle morphology, melting point and suitability for milling
• Characterizations of importance for the dosage forms of choice, e.g. bulk density, powder flow, angle of repose and compression properties.
Theoretical treatments of these molecular properties, and laboratory methods for measuring them, which are beyond the scope of this book, are described in textbooks such as Burger and Abraham (2003), Aulton (2007) and Allen (2008). Here we consider some issues that commonly arise in drug development.
Solubility and dissolution rate
The question of solubility, already emphasized in Chapters 9 and 10, is particularly important in relation to the development of pharmaceutical formulation. It is measured by standard laboratory procedures and involves determining the concentration of the compound in solution after equilibration – usually after several hours of stirring – with the pure solid. In general, compounds whose aqueous solubility exceeds 10 mg/mL present no problems with formulation (Kaplan, 1972). Compounds with lower solubility are likely to require conversion to salts, or the addition of non-aqueous solvents, in order to achieve satisfactory oral absorption. Because the extreme pH values needed to induce ionization of very weak acids or bases are likely to cause tissue damage, the inclusion of a miscible solvent of relatively low polarity, such as 20% propylene glycol or some other biocompatible solubilizing agent (see below), will often be required for preparing injectable formulations. Complications may arise with oral formulations if the solubility is highly dependent on pH, because of the large pH difference between the stomach and the small intestine. Gastric pH can range from near neutrality in the absence of any food stimulus to acid secretion, to pH 1–2, whereas the intestinal pH is around 8. Basic substances that dissolve readily in the stomach can therefore precipitate in the intestine and fail to be absorbed. Compounds that can exist in more than one crystal form can also show complex behaviours. The different lattice energies of molecules in the different crystal forms mean that the intrinsic solubility of the compound is also different. Different crystal forms may correspond to different hydration states of the compound, so that a solution prepared from the unhydrated solid may gradually precipitate as hydrated crystals. Selecting the best salt form to avoid complications of this sort is an important aspect of preformulation studies.
In practice, dissolution rates depend mainly on:
• Intrinsic solubility (since this determines the boundary layer concentration)
• Molecular weight (which determines diffusion coefficient)
• Particle size and dispersion of the solute (which determine the surface area of the boundary layer and the length of the diffusion path).
Routes of administration and dosage forms
With the knowledge of the characteristics of the drug substance from preformulation studies, the administration route has to be selected and the substance to be included into a dosage form which is effective and convenient for the patient. The preferred dosage form for therapeutic agents is almost always an oral tablet or capsule, either taken as needed to control symptoms, or taken regularly once or several times a day. However, there are many alternatives, and Table 16.1 lists some of the main ones. An important consideration is whether it is desirable to achieve systemic exposure (i.e. distribution of the drug to all organs via the bloodstream) or selective local exposure (e.g. to the lungs, skin or rectum) by applying the drug topically. In most cases systemic exposure will be required, and an oral capsule or tablet will be the desired final dosage form. Even so, an intravenous formulation will normally be required for use in safety pharmacology, toxicology and pharmacokinetic studies in man.
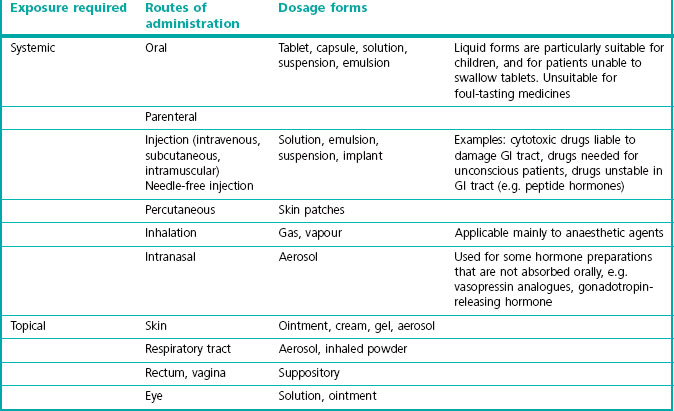
Transdermal administration of drugs formulated as small adhesive skin patches has considerable market appeal, even though such preparations are much more expensive than conventional formulations. To be administered in this way, drugs must be highly potent, lipid soluble and of low molecular weight. Examples of commercially available patch formulations include nitroglycerin, scopolamine, fentanyl, nicotine, testosterone, estradiol and ketoprofen, and several others in development. The main limitation is the low permeability of the skin to most drugs and the small area covered, which mean that dosage is limited to a few milligrams per day, so only very potent drugs can be given systemically via this route of administration. Variations in skin thickness affect the rate of penetration, and the occurrence of local skin reactions is also a problem with some drugs. Various penetration enhancers, mainly surfactant compounds of the sort discussed above, are used to improve transdermal absorption. The transfer rate can be greatly enhanced by applying a small and painless electric current (about 0.5 mA/cm2), and this is effective in achieving transfer of peptides (e.g. calcitonin) and even insulin through the skin. It also offers promise as a route of administration of oligonucleotides in gene therapy applications (see Chapter 12). These procedures are used for administration of nucleotides and in gene therapy and are being used experimentally in the clinic, but are not yet available as commercial products for routine clinical use. Ultrasonic irradiation is also under investigation as a means of facilitating transdermal delivery. These procedures would also allow the administration to be controlled according to need. Intranasal drug administration (Illium, 2002, 2003) is another route that has been used successfully for a few drugs. The nasal epithelium is much more permeable than skin and allows the transfer of peptide drugs as well as low-molecular-weight substances. Commercially available preparations have been developed for peptide hormones, such as vasopressin analogues, calcitonin, buserelin and others, as well as for conventional drugs such as triptans, opioids, etc. The main disadvantages are that substances are quickly cleared from the nasal epithelium by ciliary action, as well as being metabolized, and the epithelial permeability is not sufficient to allow most proteins to be given in this way. Ciliary clearance can be reduced by the use of gel formulations, and surfactant permeability enhancers can be used to improve the penetration of larger molecules. The possibility of administering insulin, growth factors or vaccines by this route is the subject of active research efforts. Some studies have suggested (see Illium, 2003) that substances absorbed through the nasal epithelium reach the brain more rapidly than if they are given intravenously, possibly bypassing the blood–brain barrier by reaching the CNS directly via the nerves to the olfactory bulb.
Oromucosal delivery (Madhav et al., 2009) and especially utilizing the buccal and sublingual mucosa as the absorption site is a drug delivery route which promotes rapid absorption and almost immediate pharmacological effect. The sublingual mucosa, especially, is highly vascularized and this route bypasses the gastrointestinal tract and, thus, the first-pass metabolism. However, not all drugs can be efficiently absorbed through the oral mucosa because of physicochemical properties or enzymatic breakdown of the drug and the amount of drug that could be absorbed is limited to a few milligrams. The drug has to be soluble, stable and able to easily permeate the mucosal barrier at the administration site. Also some formulation aspects have to be taken into consideration, using a tablet formulation for a rapid onset of effect a prerequisite is a fast disintegration and dissolution in the oral cavity resulting in an optimal exposure of active substance to the small volume of dissolving fluids. Swallowing of the drug could be a potential problem, but can be minimized by using technologies using mucoadhesive components (Bredenberg et al., 2003; Brown and Hamed, 2007). An optimized formulation and using this administration route has the potential for very fast absorption (Kroboth et al., 1995) and obtaining peak blood levels within 10 to 15 minutes. It is thereby potentially a more comfortable and convenient alternative to the intravenous route of administration.
Formulation
• Chemical stability (including stability at low pH)
• Permeability across the gastrointestinal epithelium
• Good access to site of action (e.g. blood–brain barrier penetration, if intended to work in the brain)
The choice of excipients and the manufacturing process is very much dependent on the characteristics of drug substance and the desired properties of the dosage form. Drug release profile is just one aspect, while the homogeneity or uniformity of content is another. If the drug substance is very potent and cohesive then mixing a small amount of drug with a high amount of filler could lead to a product with low homogeneity. This problem is most severe if the drug particles are micronized to improve the dissolution rate. Then it is important to also choose the most appropriate manufacturing process, such as dry mixing with larger filler paticles, so-called ordered or interactive mixing, wet granulation or drug coating of placebo tablets. Different processes used in drug development and manufacturing are described in textbooks such as Aulton (2007).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


