(1)
Department of Biotechnology & Biophysics, Biozentrum, Julius-Maximilians-University Würzburg, Würzburg, Germany
Abstract
Quenching of organic fluorophores by aromatic amino acids and DNA nucleotides with expelled electron donating properties allows the study of conformational dynamics of biomolecules. Efficient fluorescence quenching via photoinduced electron transfer (PET) requires van der Waals contact and can be used as reporter for structural fluctuations at the 1-nm scale in proteins, peptides, and nucleic acids. The combination of PET with fluorescence correlation spectroscopy (FCS) establishes a powerful method (PET-FCS) to study equilibrium dynamics at the single-molecule level on time scales from nano- to milliseconds. We delineate the fundamentals of PET-based fluorescence quenching, reporter engineering, instrumental and experimental design, and provide examples.
Key words
Fluorescence quenchingPhotoinduced electron transferFluorescence correlation spectroscopyProtein folding and dynamicsIntrinsically disordered proteins1 Introduction
1.1 General Introduction
The ability of polypeptides and nucleic acids to spontaneously adopt unique three-dimensional folds in aqueous solution is one of the most fundamental examples of biological self-assembly. Following the traditional structure-function relationship, function is determined by the exact three-dimensional arrangement of building blocks, i.e., amino acids and nucleotides. The discovery of proteins that natively lack regular structure, so-called intrinsically disordered proteins, challenges this traditional view and makes a mechanism of function difficult to comprehend [1, 2]. An estimated 40 % of proteins in the human proteome contain intrinsic disorder, highlighting their importance in higher organisms [3]. Disordered proteins are inherently dynamic, and the role of dynamics for function has been recognized in recent years [4]. Protein conformational motions occur on broad length and time scales and are often modulated by binding.
In order to monitor motions within biopolymers, changes in distance between specific sites need to be measured as function of time. From the spectroscopic point of view, any energy transfer method such as resonant or nonresonant energy transfer, proton transfer, or electron transfer can be used to reveal conformational changes, as long as the change of spectroscopic signal results from alterations in the interaction distance or geometry of extrinsic or intrinsic probes. In this context, fluorescence spectroscopy is a method of choice because emission properties of chromophores are sensitive to their short-range and long-range environment. The exquisite sensitivity of modern fluorescence spectroscopy allows detection down to the ultimate limit of single molecules. In recent years, single-molecule (sm) fluorescence has arrived as integral part of the spectroscopic tool box for molecular biology [5, 6]. Sm methods detect heterogeneity that is hidden in the ensemble average signal of conventional spectroscopy. The approach is therefore well suited to study structural changes of proteins and nucleic acids, molecules that can be considered dynamically heterogeneous in structural terms. The perhaps most popular and applied fluorescence reporter system for distance changes at a molecular scale is fluorescence resonance energy transfer (FRET) [5, 7–9]. FRET involves the radiationless transfer of excited-state energy from a donor (D) to an acceptor (A) fluorophore, a photophysical process that occurs through space via dipole-dipole coupling. The FRET signal scales with 1/(1 + (R/R 0)6), where R 0 is the so-called Förster radius of 50 % transfer efficiency and ranges typically between 2 and 8 nm, depending on the photophysical properties of D and A as well as environmental parameters. In the case of smFRET, however, R 0 is typically on the order of 5 nm, due to the stringent demands on brightness restricting the selection of fluorophores. Thus, smFRET monitors distance changes typically around 5 nm.
Photoinduced electron transfer (PET), on the other hand, is a photochemical process that quenches fluorescence emission through charge transfer between fluorophore (F) and quencher (Q) at van der Waals contact [6, 10, 11]. Contact typically occurs at <1 nm distance separation between the geometric centers of F and Q [12]. But PET fluorescence quenching has also energetic constraints. Only if the reduction and oxidation potential of F and Q match, the fluorescence-quenching electron transfer can occur spontaneously. Among all natural occurring building blocks of proteins and nucleic acids, it is only the amino acid tryptophan (Trp) and the nucleobase guanosine (G) that have oxidation potentials low enough to serve as efficient electron donors in PET reactions with extrinsic labels. This unique redox property makes them an exquisite tool to site-specifically transform protein and nucleic acid conformational changes into on/off fluorescence fluctuations of labels placed at specific sites within a structure. When observed in highly dilute solution on confocal fluorescence microscopes, structural fluctuations emerge as fluorescence fluctuations from individual molecules and can be analyzed by correlation spectroscopy. In recent years, PET in combination with fluorescence correlation spectroscopy (PET-FCS) has arrived as valuable tool to study fast conformational dynamics of peptides [13–15], proteins [16–20], and nucleic acids [21, 22].
In the following two subsections, we introduce the basics of PET and its combination with FCS.
1.2 Photoinduced Electron Transfer (PET) Reactions
Fluorophores are capable of reporting changes in their local environment. Dependent on the photophysical properties of the fluorophore, changes in the microenvironment are accompanied by alterations in fluorescence characteristics like spectrum, quantum yield, or lifetime. If excitation occurs through absorption of light in a fluorophore (usually an organic molecule with a delocalized aromatic π-electron system), its reactivity generally increases enabling redox reactions that are energetically forbidden in the singlet ground state. In PET experiments, a single fluorophore and a quencher are incorporated into the biopolymer chain. Depending on oxidation and reduction potentials, electron transfer from the fluorophore, being the donor D, to its interaction partner, the acceptor A, or vice versa can occur, resulting in non-radiative dissipation of excited-state energy. In order to analyze and interpret experimental results from fluorescence quenching caused by PET, the transfer mechanism and its distance dependence has to be well understood. In general, quenching of fluorophores in the first excited singlet state by electron donors or acceptors results in charge separation and the formation of a radical ion pair 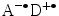 , which returns to the ground state via charge recombination (Fig. 1). The efficiency of charge separation is controlled by the relation between the free energy of the reaction, the reorganization energy, and the distance between donor and acceptor.
, which returns to the ground state via charge recombination (Fig. 1). The efficiency of charge separation is controlled by the relation between the free energy of the reaction, the reorganization energy, and the distance between donor and acceptor.
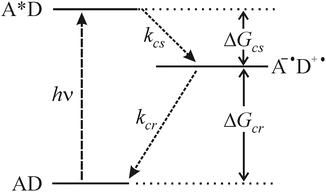
, which returns to the ground state via charge recombination (Fig. 1). The efficiency of charge separation is controlled by the relation between the free energy of the reaction, the reorganization energy, and the distance between donor and acceptor.Fig. 1
Kinetic and thermodynamic scheme of PET charge separation and charge recombination. A represents the acceptor fluorophore, and D is the electron donor, e.g., Trp. (k cs: rate constant for charge separation; k cr: rate constant for charge recombination; ΔG cs: free energy change for charge separation; ΔG cr: free energy change for charge recombination)
To estimate the efficiency of photoinduced charge separation, the associated change in free energy, ΔG cs, can be estimated using the Rehm-Weller equation [23]
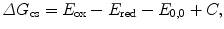
where E ox and E red are the first one-electron oxidation potential of the donor and the first one-electron reduction potential of the acceptor, respectively, in solvent. E 0,0 is the energy of the zero-zero transition to the lowest excited singlet state, and C is the solvent-dependent Coulombic interaction energy (which can be neglected in moderately polar environment). The reaction rate follows an exponential D-A distance dependence with a characteristic length scale on the order of few Angström [24]. Evidence for PET reactions can be derived from experiments showing the transient existence of charged-separated species. Measured redox potentials allow the estimation of PET efficiencies based on the Rehm-Weller formalism (Eq. 1) and strongly support the photochemical process.
(1)
The kinetics of PET reactions between electron donors like Trp or G and fluorophores are ultrafast, i.e., in the femtosecond (fs) to picosecond (ps) time regime [25, 26]. Examples for PET between fluorophores and planar aromatic ring systems, such as the indole moiety of Trp or the fused pyrimidine-imidazole ring system present in G, including redox potentials can be found in the literature [11, 27–29]. The oxidation potentials of Trp (E ox = 0.60–1.00 V versus SCE) and guanine (E ox = ~1.24 V versus SCE) are unique among all naturally occurring amino acids and nucleobases. In combination with suitable fluorophores, Trp and G in polypeptides and oligonucleotides serve as selective electron donors in PET reactions. The oxazine derivative MR121, for instance, has a reduction potential of E red = −0.42 V vs. SCE (measured in acetonitrile) and a transition energy of ~1.9 eV, such that PET from Trp or G to MR121 is exergonic (ΔG cs between −0.7 and −0.2 eV) according to Eq. 1. Note that commercial oxazine fluorophores, like ATTO 655 or ATTO Oxa11, have the chromophore of MR121. Besides oxazine derivatives, rhodamine and BODIPY fluorophores are also prone to being efficiently quenched by electron donors [11].
Interactions between fluorophore and quencher can either be “dynamic” or “static.” Whereas collisional quenching of fluorophores by suitable compounds (electron donors or acceptors) is reflected in a decrease in fluorescence lifetime and intensity (“dynamic quenching”), complex formation between the fluorophore and quencher leads to efficient “static quenching” without observed changes in fluorescence lifetime. These molecular complexes feature stacked interaction geometries [12] that are essentially nonfluorescent and dissociate much slower than the fluorescence decay time. Thus, association and dissociation of fluorophore and quencher leads to digital off/on fluorescence switching, the kinetics of which depend on the microscopic rate constants of complex formation and dissociation.
The formation of efficiently quenched fluorophore/quencher complexes in aqueous solution is the basis for using them as reporter for rapid conformational fluctuations. Through site-specific attachment of fluorophore and quencher, distance variations between chosen attachment points can be observed spectroscopically and analyzed temporally.
1.3 Combining PET with Fluorescence Correlation Spectroscopy
Fluorescence correlation spectroscopy (FCS) analyzes the kinetics of fluorescence fluctuations at equilibrium through statistical analysis of intensity time traces. Virtually any process that gives rise to a fluctuation of fluorescence emission can be analyzed via FCS. With the advent of modern fluorescence microscopy techniques and equipment within the past decade, FCS has been integrated as single-molecule technique for molecular biology. One main application of classical FCS is to measure the time constants of molecular diffusion, a quantity that is related to the size and shape of a molecule as well as interactions within its environment. The technique can thus provide information on biomolecular structure and function. For details on FCS and its applications, the reader is referred to reviews [30–32].
In FCS fluorescence intensity time traces are recorded from molecules diffusing through a spatially well-defined excitation/detection volume by Brownian motion. Typically, micro-ensembles of molecules are probed from pM to nM samples, yielding about 1–100 molecules at a time in the detection focus depending on microscope setup geometry. Fluorescence fluctuations are analyzed by calculating the autocorrelation function (ACF), G(τ), of the recorded fluorescence intensity time trace:
![$$ G(\tau )=\frac{{< I(t)\cdot I(t+\tau )}>}{{I{(t)^2}}}, $$
” src=”/wp-content/uploads/2017/03/A299540_1_En_27_Chapter_Equ00272.gif”></DIV></DIV><br />
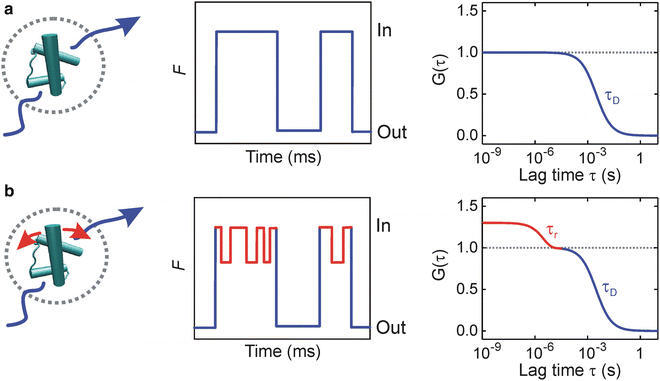
<DIV class=EquationNumber>(2)</DIV></DIV>where <SPAN class=EmphasisTypeItalic>I</SPAN>(<SPAN class=EmphasisTypeItalic>t</SPAN>) is the fluorescence intensity at time <SPAN class=EmphasisTypeItalic>t</SPAN>, <SPAN class=EmphasisTypeItalic>I</SPAN>(<SPAN class=EmphasisTypeItalic>t</SPAN> + <SPAN class=EmphasisTypeItalic>τ</SPAN>) is the fluorescence intensity after a time lag <SPAN class=EmphasisTypeItalic>τ</SPAN>, and < > denotes the time average over the total observation time. Under appropriate experimental conditions, constantly emitting fluorescent labels give rise to a stochastic fluctuation of the detector signal as molecules diffuse in and out of the focal volume (Fig. <SPAN class=InternalRef><A href=]() 2). This process results in a decay of the ACF with a characteristic time constant τ D representing the experimental diffusion time constant. τ D is typically on the order of one ms and depends, besides on the size of the observation volume, on the size and shape of the molecule, temperature, solution viscosity, and specific interactions within the diffusion matrix.
2). This process results in a decay of the ACF with a characteristic time constant τ D representing the experimental diffusion time constant. τ D is typically on the order of one ms and depends, besides on the size of the observation volume, on the size and shape of the molecule, temperature, solution viscosity, and specific interactions within the diffusion matrix.
Get Clinical Tree app for offline access
Fig. 2
Probing protein dynamics by PET-FCS. Fluorescently modified proteins diffuse through a confined detection volume by Brownian motion, causing a stochastic fluctuation of the fluorescence signal (F) at the detector. (a) A protein that shows constant fluorescence emission gives rise to fluctuations that result in a decay of the ACF (G(τ)) with a characteristic time constant of translational diffusion τ D (blue). (b) Kinetics of conformational motions that are faster than τ D and modulate fluorescence emission of the extrinsic label emerges as additional decay in the ACF with an observed relaxation time constant τ r (red) (Adapted with permission from ref. 20. Copyright 2011, Elsevier)
Through incorporation of a suitable fluorophore and Trp or G as quenchers at specific sites within a biomolecule, conformational changes that induce fluorophore/quencher association and dissociation are transformed into changes of fluorescence emission intensity. If equilibrium conformational fluctuations occur on time scales faster than τ D, corresponding kinetics emerge as additional decay in the ACF with an observed relaxation time constant τ r (Fig. 2). This represents the basic idea of a PET-FCS experiment.
Assuming two-state equilibrium between fluorescent and fluorescence-quenched conformational states A and B, the observed relaxation time constant τ r contains the microscopic rate constants k 1 and k −1 (Scheme 1): τ r = 1/(k 1 + k −1). Deconvolution requires knowledge of the equilibrium constant K eq = k 1/k −1, which may be obtained from independent experiments. The amplitude a r of the ACF decay contains K eq but is convoluted with the change of fluorescence brightness associated with the transition from state A to B. The quenched state B may, e.g., be an ensemble of states showing residual fluorescence that results from rapid conformational dynamics of the chain segment separating fluorophore and quencher. B may therefore be a dim state, in which case K eq does not equal a r.
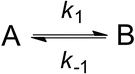
Scheme 1
Two-state equilibrium of conformational transition between states A and B
PET-FCS is particularly useful for the measurement of kinetics of loop closure within unfolded polypeptides or single-stranded nucleic acids. Loop closure is an early event in folding, and the underlying kinetics is a useful parameter to investigate structure and dynamics of denatured states or intrinsically disordered proteins. Intra-chain complex formation of fluorophore and Trp or G within an unfolded polypeptide or nucleic acid closes a loop and results in off-switching of fluorescence emission. The underlying kinetics can be described as in Scheme 2.
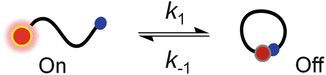
Scheme 2
Two-state equilibrium of loop closure
For loop closure, the microscopic rate constants can be calculated from τ r and a r because the closed fluorescence-quenched configuration is assumed to be a true off-state that results from a stacked complex between fluorophore and quencher. In this case, a r equals the equilibrium constant K eq of loop closure.
2 Materials
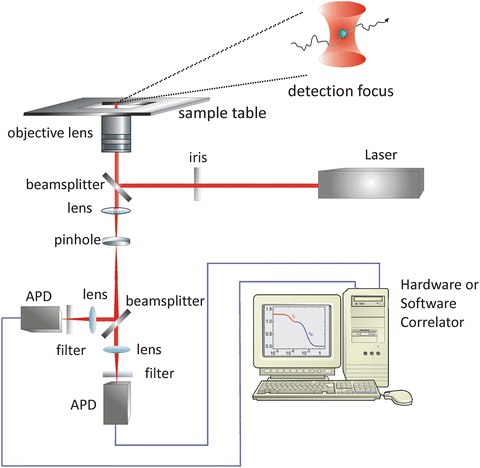
2.1 The PET-FCS Microscope
A typical microscope setup for PET-FCS experiments is illustrated in Fig. 3. The basic components are an inverted fluorescence microscope body equipped with an objective lens, a continuous-wave laser as excitation source, two avalanche photodiodes (APDs) as detection devices, a digital hardware correlator or PC plug-in card for data acquisition plus correlation software, and optical elements. The wavelength of the laser light is selected to match the electronic absorption band of the fluorophore (see Note 1). The laser should feature a well-defined (ideally spherical) beam profile, as well as temporally stable emission intensity. The laser light beam is guided into the back aperture of the microscope body via optical mirrors. The laser light power at the back aperture of the microscope can be conveniently adjusted using an optical density filter positioned between laser and microscope. The laser light beam profile can be shaped and size-adjusted using an iris mounted to the back aperture of the microscope body (see Note 2). Suitable objective lenses have oil immersion medium, a 60× magnification, and high numerical aperture (e.g., NA 1.4 or higher). Fluorescence light from the sample is collected by the same objective lens and passed through a dichroic beam splitter that separates fluorescence from excitation light. The fluorescence light leaves the microscope body and is guided by mirrors onto a 50 % nonpolarizing cubic beam splitter that shares the light equally between two single-photon sensitive APDs. Band-pass or long-pass optical filters are placed between the exit of the microscope body and the cubic beam splitter to eliminate residual background from excitation light. A lens positioned in the fluorescence light path is used to focus the fluorescence light onto the active zone of APDs. In practice, fiber-coupled APDs equipped with 100-μm optical fibers are recommended as detector devices because they are (1) convenient to adjust in the fluorescence light path using x, y, z optical stages and (2) serve as spatial filter for the fluorescence light before it passes onto the photodiode, thereby replacing the pinhole in conventional confocal setups. Alternatively, the setup may be equipped with a 100-μm pinhole as spatial filter in the detection path. Two APDs are required in order to bypass their dead time and after-pulsing effects through cross-correlation of detector signals yielding a pseudo-ACF (see Note 3). Cross-correlation can either be performed by recording the detector signals individually using a PC plug-in card, followed by software correlation, or directly by using a digital hardware correlator device in cross-correlation mode. The advantage of hardware correlation is that the evolution of the ACF can be directly observed in real time, alleviating troubleshooting during data acquisition. Further, the size of an ACF output file from a hardware device is small (typically <1 MB), while the time trace recorded over a macroscopic time window for subsequent software correlation can easily exceed 1 GB. The time resolution of modern hardware correlators is on the order of 1 ns, i.e., fast enough to capture most biologically relevant time scales. The disadvantage of hardware correlation, however, is that the raw fluorescence intensity time traces are lost since they are analyzed on the fly and that the lag-time window to calculate the ACF cannot be varied.