FIGURE 67-1 The 12-lead ECG recorded in the MICU showing complete heart block and QTc prolongation.
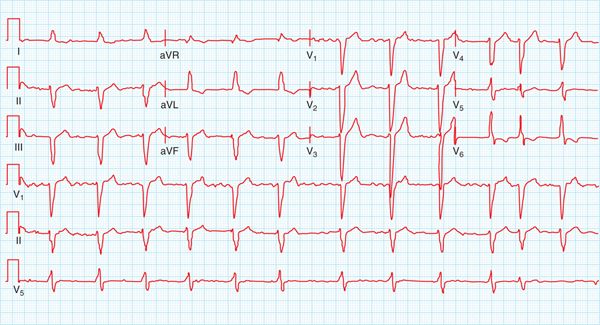
FIGURE 67-2 The 12-lead ECG recorded at prior visit showing LBBB and a QTc of 450 ms. In the presence of LBBB, this is a normal QTc.
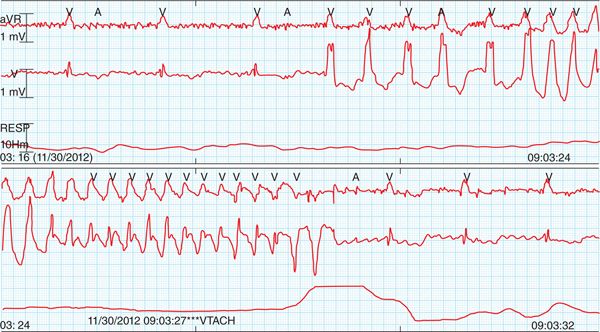
FIGURE 67-3 Telemetry strip showing onset and offset of TdP.
CASE EXPLANATION
The patient presented with recurrent syncope complicated by a basilar artery thrombosis. While it is impossible to be certain, it seems likely that the syncope was caused by either the onset of complete heart block or the development of TdP, and the onset of coma appears to be attributable to the basilar artery occlusion. While the etiology of the complete heart block is likewise uncertain, the presence of preexisting left bundle branch block suggests significant infra-His conduction disease. The manifestation of a new right bundle branch block and regularization of the ventricular response is most consistent with complete atrioventricular block. The etiology of the polymorphic ventricular tachycardia is not mysterious, however, and is due to the new onset of severe bradycardia and resultant QT interval prolongation associated with the escape rhythm. The immediate and permanent resolution of the arrhythmia with the institution of ventricular pacing, the absence of a history of QT prolongation, and the lack of a QT-prolonging drug indicate that this is pause-dependent TdP, although some contribution from the cerebral insult cannot be ruled out.
EPIDEMIOLOGY
• The prevalence of true pause-dependent TdP is unknown but is likely rare. Sporadic case reports document QT prolongation and TdP in children and adults after congenital or acquired complete heart block.1–3
 In its “pure” form, it is associated with a normal QT interval prior to the pause.
In its “pure” form, it is associated with a normal QT interval prior to the pause.
• TdP commonly presents as two syndromes: syncope and sudden cardiac death (SCD).
 Syncope is a common syndrome, with an incidence of 6.2 cases per 1000 person-years, with a cardiac cause identified in 9.5%, but the percentage of cases attributable to pause-dependent TdP is likely very small.4
Syncope is a common syndrome, with an incidence of 6.2 cases per 1000 person-years, with a cardiac cause identified in 9.5%, but the percentage of cases attributable to pause-dependent TdP is likely very small.4
 At autopsy, a long QT (LQT) associated gene mutation (a congenital predisposition to TdP) was identified in approximately one-third of cases of SCD without structural heart disease.5
At autopsy, a long QT (LQT) associated gene mutation (a congenital predisposition to TdP) was identified in approximately one-third of cases of SCD without structural heart disease.5
 TdP is also associated with structural heart disease, with an odd’s ratio of 3.9 in NYHA Class III or IV dilated cardiomyopathy exposed to dofetilide, a potent inhibitor of the rapid component of the delayed rectifier potassium current, IKr.6
TdP is also associated with structural heart disease, with an odd’s ratio of 3.9 in NYHA Class III or IV dilated cardiomyopathy exposed to dofetilide, a potent inhibitor of the rapid component of the delayed rectifier potassium current, IKr.6
 In 60 cases collected over 11 years, the most common causes of TdP include QT-prolonging drugs (38%), hypokalemia (27%), and severe bradycardia (35%).7
In 60 cases collected over 11 years, the most common causes of TdP include QT-prolonging drugs (38%), hypokalemia (27%), and severe bradycardia (35%).7
ETIOLOGY AND PATHOPHYSIOLOGY
• Torsades de pointes (TdP) is a polymorphic ventricular tachycardia typified by continuous change in the QRS axis in the setting of a prolonged QT interval, often occurring in short bursts.
• The long QT interval can be congenital or acquired and reflects increased heterogeneity of repolarization, a necessary requirement for induction of reentrant arrhythmias.
• TdP is typically—perhaps always—multifactorial in origin, requiring interaction of depressed “repolarization reserve”8 due to drugs, structural heart disease, ischemia, electrolyte imbalances, female gender, altered calcium handling, or bradycardia combined with a trigger in the form of a premature beat (Table 67-1).
TABLE 67-1 Loss of Repolarization Reserve: Risk Factors for Torsades de Pointes
hERG mutation (0.1%-1% of population) |
QT-prolonging drug use, particularly at high dose |
Preexisting QTc >450 ms |
Electrolyte disturbances ↓ K+, ↓ Mg2+ |
Female gender |
Bradycardia |
Atrial fibrillation |
Combination of QT-prolonging drugs |
Inhibition of drug metabolism |
Cardiac ischemia/congestive heart failure |
Liver disease, eg, cirrhosis |
Anorexia nervosa/HIV |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


