2.2 STRUCTURE AND PROPERTIES OF MEMBRANES
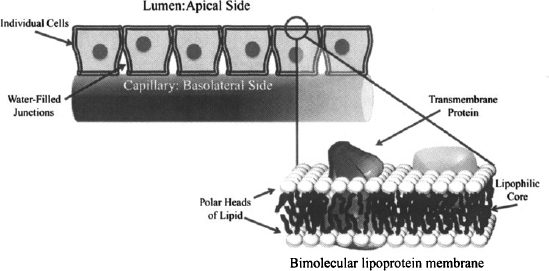
To get into the body and get taken up by the tissues, drugs have to penetrate the epithelial membranes that line the major organs and body cavities. Epithelial membranes consist of a series of cells joined by water-filled junctions (Figure 2.2). The membranes of these cells consist of a bimolecular layer of lipoproteins. The two layers of lipids are oriented such that the polar ends of the molecule point out toward the aqueous medium or the outside of the membrane, and the lipid component makes up the inner core or the matrix of the membrane. Various protein molecules are embedded at different points in the membranes (Figure 2.2). The portion of the epithelial membrane that faces the outside of the organ or body (i.e., the side facing the gastrointestinal lumen) is called the apical side. The remaining sides, including the side that faces the circulatory system, are the basolateral sides. Individual membranes may consist of a single layer or multiple layers of cells. During each process in ADME, a drug may have to pass across several membranes. For example, orally administered drugs must pass through the gastrointestinal wall into the interstitial fluid and then through the capillary membrane to enter the blood. From there they may have to pass several other membranes to access their site of action and to be removed from the body. Drugs penetrate these membranes by either passive diffusion or by a transport-mediated process.
FIGURE 2.2 Cell membrane. Individual epithelial cells are joined together by water-filled junctions. The membranes of the cells consist of bimolecular layers of lipoproteins. The polar portion of the lipid point toward the outer side of the membrane, and the lipid component makes up the inner core of the membrane. The apical side of the membrane points toward the outside, or lumen, and the remaining sides are known as the basolateral sides. (Diagram drawn by Linnea E. Anderson.)
2.3 PASSIVE DIFFUSION
Passive diffusion is the most common way for drugs to pass through biological membranes. The concentration gradient across a membrane is the driving force for the process, which tries to equalize the drug concentrations on either side of the membrane. As a result, there is a net movement of drug from the side with a high concentration to the side with a low concentration. Any drug that is bound to tissue macromolecules or plasma proteins is essentially taken out of circulation and does not participate in the concentration gradient. The process of diffusion is governed by Fick’s law and may be expressed as:
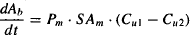
where dAb/dt is the amount of drug diffusing per unit time (mg/h), Pm the permeability of the drug through the membrane (cm/h), SAm the surface area of the membrane (cm2) Cu1 the higher unbound drug concentration (mg/mL), and Cu2 the lower unbound concentration (mg/mL).
In most situations (e.g., during drug absorption and the initial phases of drug distribution), drug that diffuses across the membrane is diluted into a very large volume. Thus, Cu2 can be considered to be considerably smaller that Cu1:

Thus, equation (2.1) may be written
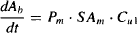
From equation (2.2) it can be seen that under these circumstances, the rate of diffusion approximates a first-order process:
(2.3) 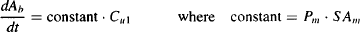
The rate of diffusion is proportional to the driving force for the process: that is, the concentration of drug. The constant of proportionality is the product of a drug’s permeability (the relative ease with which the drug passes the membrane) and the surface area of the membrane.
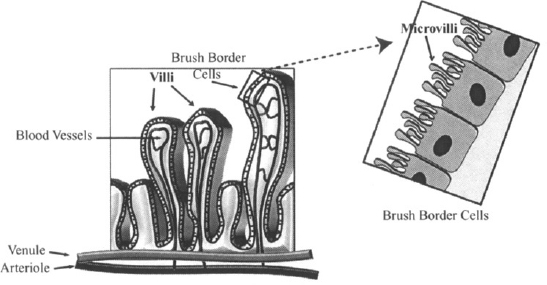
Surface Area According to equation (2.2), the rate of diffusion increases as a membrane’s surface area increases. The importance of surface area is illustrated by considering the structure of the membrane of the small intestine, a tissue whose function is to absorb essential nutrients from digested foods. To assist in this function, the membrane of the small intestine is folded into villi, or fingers, which are estimated to increase the surface area of the small intestine about 10-fold. Each of the villi is folded further into microvilli (Figure 2.3), which are estimated to increase the surface area an additional 20-fold. This extremely large surface area greatly enhances the absorptive properties, a feature that can also be taken advantage of by drugs administered orally. The very favorable absorption of the small intestine results in its being the primary site for the absorption of drugs taken orally.
FIGURE 2.3 Villi and microvilli in the small intestine. The membrane of the small intestine is folded into villi, which are then folded further into microvilli. This provides the membrane with a very large surface area, ideally suited for the absorption of nutrients and drugs. (Diagram drawn by Linnea E. Anderson.)
Permeability Permeability reflects the ability or speed with which a drug can pass through a membrane. It is dependent on both the characteristics of the membrane and the physicochemical properties of the drug: specifically, lipophilicity, charge, and size. The impact of these variables on diffusion depends on whether a drug passes through the membrane by the transcellular route or by the paracellular route. In transcellular transport, drugs diffuse through the matrix or core of the membrane. In paracellular transport, drugs diffuse through the water-filled gaps between adjacent cells (Figure 2.4).
FIGURE 2.4 Transcellular and paracellular diffusion. Passive diffusion is the most common way that drugs penetrate membranes. They can pass though the matrix of the cell (transcellular passage) or through the water-filled junctions between adjacent cells (paracellular transport). (Diagram drawn by Linnea E. Anderson.)
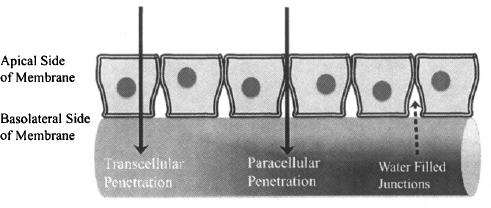
The nature of the core of the membrane is essentially constant from one type of membrane to another. As a result, the principles of transcellular diffusion are the same for all membranes, including the gastrointestinal membrane, the blood–brain barrier, and the renal tubular membrane. Paracellular diffusion is controlled by the nature of the junctions between adjacent cells of the membrane. These vary from tissue to tissue. As a result, and in contrast to transcellular diffusion, the ability of a drug to pass through a membrane by the paracellular route will vary from one tissue to another.
2.3.1 Transcellular Passive Diffusion
The ease of transcellular diffusion is determined by a drug’s permeability across the lipophilic matrix of the membrane. As such, it depends on the lipophilicity, polarity, and size of the drug molecule. A drug’s lipophilicity is probably the most important determinant of permeability. A drug’s lipophilicity, or fat-loving nature, is traditionally assessed by measuring its distribution between the immiscible phases of n-octanol and water. The ratio of the drug’s concentration in n-octanol and water is the drug’s partition coefficient (P):
(2.4) 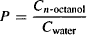
Because of the very wide range of P values among therapeutic drugs, P values are expressed most conveniently on a log scale. Table 2.1 provides the log P values of some therapeutic drugs.
TABLE 2.1 Calculated Values of Log P, Log D7.4, and Log D6.0 of Several Drugs
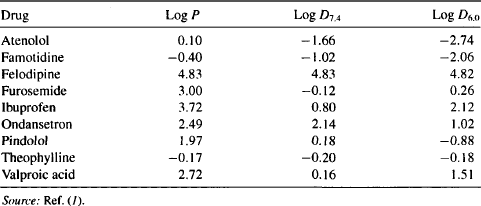
Drugs with large positive log P values (felodipine) preferentially partition into the lipid layer. They are lipophilic, would have high permeability across the lipophilic core of the membrane, and would be expected to diffuse easily. As the log P value decreases among a series of drugs, lipophilicity and permeability both decrease and transcellular membrane penetration would become increasingly difficult. Drugs with very low or negative log P values (e.g., atenolol and famotidine) partition primarily into the aqueous phase, are more polar in nature, and would be expected to have poor membrane permeability.
The disadvantage of the partition coefficient is that it measures the distribution of drugs between the two phases when the drug is completely in the nonionized state. Thus, it is a measure of a drug’s inherent or intrinsic lipophilicity. But most drugs are either weak acids or bases and, as a result, exist in biological fluids in equilibrium between their ionized and nonionized forms. For example, a drug that is a weak acid would ionize as follows in an aqueous medium:
(2.5) 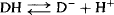
The degree of ionization will influence a drug’s permeability because only the nonionized form of the drug would be able to penetrate a lipophilic membrane. A more useful measure of membrane permeability is the partition coefficient measured at a specific, biologically relevant pH. The partition coefficient of a drug between a lipid phase and an aqueous phase at a Specific pH is referred to as the distribution coefficient (D), or log D on the log scale. The log D7.4 and log D6.0 values of several drugs are shown in Table 2.1. It can be seen that a drug’s log D value is generally less than its log P value, which reflects the fact that when the drug is partially ionized, the ionized form cannot partition into the lipid phase, which makes the drug effectively less lipophilic than is the totally nonionized form of the drug. It can also be seen that the log D values of drugs that are weak acids (e.g., ibuprofen, furosemide, valproic acid) are higher at the more acidic pH of 6.0 than at pH 7.4. This reflects the fact that drugs are less ionized at more acidic pH’s. Conversely, the log D values of drugs that are weak bases (e.g., atenolol, ondansetron) are lower at a more acidic pH of 6.0, owing to the greater degree of ionization.
The size or mass of the drug molecules also affects the permeability, as large molecules experience difficulty diffusing. Studies suggest, however, that mass does not appear to be important if it is below about 400 daltons (Da), which includes many of the drugs presently used therapeutically. Transcellular diffusion has been studied most extensively in the context of the absorption of orally administered drugs. Pharmaceutical companies want to maximize the possibility that newly developed drugs can be given orally, and much research has been conducted to try to identify critical physiochemical properties of drugs. Studies have emphasized the importance of a high log D value. These studies also indicate that mass and polarity are important and suggest that drugs with a molecular mass greater than 500 Da are likely to have poor membrane permeability, particularly if they also possess additional adverse physicochemical characteristics, such as polarity or poor lipophilicity (2).
In summary, transcellular permeability is highest for small lipophilic, nonpolar drugs.
2.3.2 Paracellular Passive Diffusion
Paracellular transport involves to the passage of drugs through the junctions between the cells of the membrane (Figure 2.4). It is dependent on the size of the junction and on the size of the drug molecule. The junctions between adjacent cells of the epithelium membrane vary from one tissue to another. The junctions between the cells in the gastrointestinal membrane and skin are very tight and serve to hold transcellular proteins in place and also, presumably, to protect the body from the penetration of foreign substances across these outside membranes. As a result, paracellular diffusion of drugs across the intestinal membrane is a very minor route of absorption. Atenolol [molecular weight or molecular mass (MW) = 266 Da; log D6.5 = -2] and terbutaline (MW = 180 Da; log D6.5 = -1.3) were thought to be absorbed by this route. However, recent evidence has cast doubt on this (3) and suggests that they may be too large for paracellular diffusion across the intestinal membrane. The aminoglycoside antibiotics, which are both too polar (log D7.4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


