and FJE Vajda2
(1)
Clinical Neurology and Neuropharmacology, University of Queensland, and Honorary Consultant Neurologist, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
(2)
Department of Medicine and Neurology Director of the Australian Epilepsy and Pregnancy Register, University of Melbourne and Royal Melbourne Hospital, Melbourne, Australia
Abstract
This chapter attempts to discuss the published information concerning the risks of structural malformations occurring in foetuses exposed to those individual antiepileptic drugs that have been widely enough used in women with epilepsy for relevant data to be available. There appears to be persuasive evidence that valproate is a dose-related teratogen, with certain malformations such as neural tube defects seeming to be particularly associated with high maternal dosage of the drug. Evidence also points towards topiramate being a dose-related teratogen. On the basis of the available data the possibility that the remaining currently used drugs are teratogens cannot be excluded, but there appears to be enough evidence to indicate that any teratogenic hazard arising from exposure to them is quite small.
As will be apparent from the contents of the previous chapter, in the earlier years of the study of associations between intrauterine antiepileptic drug exposure and foetal malformations, there was a tendency for authors to write as if the malformations represented an unwanted class effect of the drugs. With the growth of knowledge as time passed, and as additional effective antiepileptic drugs became available, it became increasingly recognised that the degree of malformation hazard varied considerably between the individual agents. The present chapter attempts to discuss the information that is available concerning the individual antiepileptic drugs that have enjoyed some use in pregnancy in recent years.
Wlodarczyk et al. (2012) have provided a reasonably contemporary and thorough review of the literature concerning individual antiepileptic drugs and foetal malformations. Their paper includes a number of useful tabulations of previously published data. No attempt is made in the present chapter to reproduce all of the Wlodarczyk et al. material, which is a valuable source of information.
Phenobarbitone and Its Congeners
Phenobarbitone was among the antiepileptic drugs that featured in the early reports of a possible association between these agents and foetal malformations, but the drug was not singled out in these reports for a culprit role. However, after finding congenital abnormalities in four of the offspring of 61 mothers who had been treated with phenobarbitone to try to prevent threatened abortions, Wheatley (1963) raised the possibility that the drug might be a teratogen . A few years later, Nelson and Forfar (1971) studied the possible association between various drugs and foetal malformations and noted that significantly more mothers than would have been expected had been taking phenobarbitone. In the earlier literature, there were other data concerning the outcomes of pregnancies exposed to phenobarbitone in women with epilepsy, but there was little explicit analysis of the phenobarbitone-related foetal hazard in its own right. Staples (1972) concluded that, up to his time of writing, there was no definite evidence incriminating the drug as a teratogen. Shapiro et al. (1976) also took the view that the drug did not cause dysmorphogenesis .
In their analysis of an international database of foetal malformations, Arpino et al. (2000) detected an association between exposure to phenobarbitone during pregnancy and the presence of oral clefts . Then Holmes et al. (2004) found a 6.5 % malformation rate in 77 offspring of mothers exposed to phenobarbitone therapy during pregnancy. This was more than four times the risk in an antiepileptic drug-unexposed control population. In a later analysis of an expanded population from the same source, Hernandez-Diaz et al. (2012) reported a 5.6 % increase in the malformation risk in 199 pregnancies in which phenobarbitone was used in antiepileptic drug monotherapy, compared with the rate in 51 antiepileptic drug-unexposed pregnancies. This increased risk was statistically significant. However, there was no evidence of a correlation between the phenobarbitone dose and the malformation risk. Tomson et al. (2011), in an analysis of 217 phenobarbitone-exposed foetuses whose records were contained in the EURAP register , found a statistically significant higher malformation rate when the maternal phenobarbitone dose exceeded 150 mg a day, as compared with the rate when lower doses had been used. The meta-analysis of Meador et al. (2008), which accumulated 945 pregnancies, found a phenobarbitone-associated malformation rate of 4.9 %, but there were no control data to permit comparisons being made.
Although it is possible that the same data may have appeared in more than one of the above reports, there now seems to probably be sufficient evidence to suggest that phenobarbitone is a teratogen . This knowledge may not have a great impact in Western medicine where the drug is tending to disappear from use in the age group in which pregnancy is likely, but it may have significant implications for less affluent communities globally where the drug is still in extensive and reasonably satisfactory use in treating epilepsy. No specific pattern of malformation appears to be particularly associated with exposure to the drug.
Phenytoin
There were two pregnancies which resulted in foetal malformations among 55 pregnancies exposed to phenytoin in monotherapy or polytherapy in the series of Janz and Fuchs (1964), and nine in 162 similarly exposed pregnancies in the report of Speidel and Meadow (1972). Then Monson et al. (1973) specifically addressed the question of phenytoin exposure in pregnancy and the occurrence of foetal malformations. They found a 6.1 % malformation rate associated with exposure to the drug in 98 children, as compared with a 2.5 % rate in the pregnancies of over 50,000 non-epileptic women. Dravet et al. (1992) employed logistic regression analysis to find a 2.98 times increased malformation rate (95 % C.I. = 1.48, 6.05) in first trimester phenytoin-exposed foetuses, compared with a control group of 117,183 pregnancies.
Since those early reports, there has been relatively little further statistically significant evidence that phenytoin possesses any significant capacity for producing teratogenesis . When Wlodarczyk et al. (2012) reviewed the existing literature, they found nine relevant studies in which the drug had been used in monotherapy during pregnancy. In eight of these studies, the relative risk or odds ratio in favour of the drug being a teratogen exceeded 1.0, but in none to a statistically significant extent. In the most recent analysis of the Australian Pregnancy Register data, there was a foetal malformation rate of 2.4 % in 41 pregnancies exposed to phenytoin monotherapy, a relative risk of 1.49 (95 % C.I. = 0.30, 7.42) compared with the foetal malformation risk in women with epilepsy who did not take antiepileptic drugs in at least the early part of pregnancy. Logistic regression analysis of malformation risk on phenytoin dose in the Australian data showed a far from statistically significant trend for the risk to decrease rather than increase with increasing drug dose. From a statistical viewpoint, overall there seems to be little convincing recent evidence that phenytoin is an important human teratogen, though the possibility that it may be responsible for occasional foetal malformations cannot be excluded.
Despite this, over the years a substantial number of authors have written about a ‘foetal hydantoin syndrome ’. The idea seems to have been first proposed in print by Hanson and Smith (1975) after authors such as Loughnan et al. (1973), Hill et al. (1974), Danks et al. (1974) and Barr et al. (1974) had observed an increased frequency of digital hypoplasia and certain mid-face alterations in babies who had been exposed to the drug during pregnancy. As originally described, the syndrome involved relatively poor intrauterine growth and development, mild mental retardation, a facial appearance which was considered characteristic, skeletal abnormalities and, in particular, digital hypoplasia . Phelan et al. (1982) considered that the full syndrome could be recognised in some 11 % of phenytoin -exposed infants and incomplete forms in another 31 %, while Andermann et al. (1982) found hypoplasia of the phalanges in 22 % of phenytoin-exposed foetuses. Nearly all of these comparatively minor abnormalities became less noticeable as the affected infants grew older. Various subsequent authors, often on the basis of single instances, attempted to add additional features to the syndrome, and one or two began to expand the concept to that of a foetal anticonvulsant syndrome. A certain amount of animal laboratory investigation attempted to explain its mechanism, while clinical argument went on regarding whether it was a genuine entity, whether it could be diagnosed reliably in the absence of knowledge of the details of antiepileptic drug exposure in pregnancy and whether its features permitted its being distinguished from the subsequently described foetal carbamazepine and foetal valproate syndromes. Kini et al. (2006) reported that experienced dysmorphologists had failed to distinguish consistently between antiepileptic drug exposed and unexposed infants in photographs of the infants’ facial features. As time has passed, interest in the matter seems to have dwindled, and the antiepileptic drug teratogenesis literature has tended to become more focussed on major and more clinically and cosmetically important malformations.
Ethosuximide
Ethosuximide is sometimes mentioned in the antiepileptic drug teratogenicity literature, but there is little indication that the drug is a significant cause of foetal malformations. The amount of information available is understandably small because nearly all the literature emanates from Western countries. In these countries, absence seizure disorders, the usual indication for the use of ethosuximide , are not often still active by the time women become pregnant, even though the disorders were active earlier in their lives. As well, because of a perhaps not adequately founded belief that using ethosuximide in monotherapy may facilitate the occurrence of convulsive seizures, the drug has often been co-prescribed with another antiepileptic drug such as phenytoin , carbamazepine or valproate . In these instances, any foetal malformations that developed, if they were not coincidental, might have been due to the co-administered drug.
Carbamazepine
There is considerably more information concerning the possible association between carbamazepine and foetal malformations than the amount that is available for the individual drugs discussed immediately above. This is probably so because carbamazepine has been in widespread use for some 40 years and is still commonly employed in Western countries in treating epilepsy in pregnant women. At a relatively early stage, the drug acquired a reputation for relative safety from the foetal point of view. This may have happened because no congenital abnormalities occurred in the pregnancies exposed to carbamazepine alone or together with other antiepileptic drugs, in the series described by Starreveld-Zimmerman et al. (1973). In that series there were 22 malformations among 247 live births to mothers who had taken other antiepileptic drugs during pregnancy.
Over the following years, various reports appeared concerning the likelihood of carbamazepine -associated teratogenesis . For instance, Kallen et al. (1989) and Kallen (1994) studied the data in the Swedish registry of malformed neonates and noted that a nonstatistically significant increase in the occurrence of spina bifida was associated with carbamazepine exposure. Lindhout and Omtzigt (1992) accepted that this particular association was a genuine one, while Little et al. (1993) reported the occurrence of a neural tube defect in a foetus that, at the age of 3 or 4 weeks of gestation, had been exposed to the high concentration of carbamazepine resulting from a single overdose of about 4800 mg, taken in a suicide attempt. Samrén et al. (1997) compared the outcome of 192 antiepileptic drug-exposed pregnancies in women with epilepsy with that in 158 untreated pregnancies. In relation to pregnancies involving carbamazepine intake, there was an increased hazard of foetal malformations (relative risk , 4.9; 95 % C.I. = 1.3, 18.0). Diav-Citrin et al. (2001) also described a statistically significantly increased hazard of foetal malformation associated with exposure to the drug during pregnancy as compared with matched and so-called ‘general’ controls (relative risk, 2.4; 95 % C.I. = 1.1, 4.56). Matalon et al. (2002), based on a meta-analysis of 1255 pregnancies, arrived at the view that carbamazepine increased the occurrence of foetal malformation rate (6.7 %) above its background value (2.34 %). Kaaja et al. (2003), in a logistic regression analysis of 740 antiepileptic drug-exposed pregnancies in women with epilepsy, found an odds ratio for carbamazepine-associated malformations of 2.5 (95 % C.I. = 1.0, 6.0) compared with the rate in 239 pregnancies not exposed to these drugs.
Thirteen of the relevant more major papers in the subsequent literature were summarised in the study of Wlodarczyk et al. (2012). These workers noted that the published major congenital malformation rates for carbamazepine monotherapy ranged between 2.2 and 6.3 %. Relative risk or odds ratio values were available for nine of the series analysed and were 1.0 or higher in eight of the nine, though statistically significant higher in only two. In the three largest series, those of Morrow et al. (2006) with 900 pregnancies, Hernandez-Diaz et al. (2007) with 873 pregnancies and Artama et al. (2005) with 805 pregnancies, the malformation rates were respectively, 2.2, 2.5 and 4.0 %.
There are a few relatively recent publications that were not included in the Wlodarczyk et al. (2012) analysis. Jentink et al. (2010) extracted from the literature a total of 2680 pregnancies in women with epilepsy exposed to carbamazepine monotherapy. There was a major congenital malformation rate of 3.3 % (95 % C.I. = 2.7, 4.2 %), with an increased spina bifida rate (odds ratio 2.6; 95 % C.I. = 1.2, 5.3) relative to that in antiepileptic drug-unexposed pregnancies. No definite association was found between carbamazepine and any other particular foetal malformation. Tomson et al. (2011), in 1402 carbamazepine-associated pregnancies drawn from the EURAP registry, found a malformation rate of 3.4 % (95 % C.I. = 1.11, 7.71 %) and produced evidence that the malformation rate was dose dependent. Holmes et al. (2011), in North American Registry data assessing major congenital malformations detected up to 3 months after the end of pregnancy, found a 2.9 % foetal malformation risk from carbamazepine exposure during pregnancy in women with epilepsy.
It seems likely that the meta-analyses and some of the literature review papers have included the same pregnancies that were previously included in other publications. Nevertheless the data seem sufficient to suggest that carbamazepine may have some capacity to be responsible for foetal malformations and probably to cause spina bifida . However, the degree of hazard appears to not greatly exceed that which applies for pregnancies in the untreated female epileptic and normal populations.
In the most recent Australian Pregnancy Register data, there were 346 pregnancies exposed to carbamazepine monotherapy, with a 5.5 % foetal malformation risk. The risk compared with that for pregnancies in women with epilepsy unexposed to antiepileptic drugs was not statistically significant (relative risk 1.68; 95 % C.I. = 0.64, 4.42). Logistic regression analysis of malformation risk on carbamazepine dose (Fig. 8.1) showed a nonstatistically significant tendency for the risk to increase with increasing dosage (P = 0.405).
The existence of a foetal carbamazepine syndrome has been described. It comprised facial dysmorphism, congenital heart defects, skeletal abnormalities , renal agenesis , ambiguous genitalia and anal atresia (Akar et al. 2012). Some of these features had been described earlier by Jones et al. (1989), notably the facial appearances and fingernail hypoplasia, together with the overall developmental delay . Ornoy and Cohen (1996) identified such a carbamazepine syndrome in six of 47 neonates exposed to the drug. Some of the arguments as to whether such appearances constitute a recognisable syndrome specific to the drug have been mentioned in relation to phenytoin (above).
Valproate
Roughly a decade after it came into clinical use in Europe, suspicion began to arise that valproate might be a human teratogen . Robert and Guibaud (1982) noted an excessive number of instances of spina bifida among 71 malformations reported in the offspring of epileptic mothers in the Rhône Valley in France and found that there was an association between this particular malformation and exposure to valproate during pregnancy (Robert and Rosa 1983). Bjerkedal et al. (1982), Lindhout and Meinardi (1984) and Lindhout and Schmidt (1986) then reported further instances of the association, while Jager-Roman et al. (1986) described the presence of major malformations in 4 of 14 foetuses (28.6 %) exposed to valproate in monotherapy. Bailey et al. (1983) mentioned that the manufacturers of valproate had at that time held data on the outcome of 33 pregnancies which had been exposed to valproate and that there were four foetal malformations, including two meningomyelocoeles, in the material. Curran (1987) reported five instances of neural tube defects in foetuses exposed to valproate in utero. Thus within the span of a few years, there were grounds for strong suspicion that exposure to valproate during pregnancy carried a heightened risk of neural tube defects occurring in the foetus.
By this time Di Liberti et al. (1984) had proposed the existence of a characteristic identifiable syndrome (the foetal valproate syndrome ) which they had recognised in all seven infants whom they had examined and who had been exposed to valproate during pregnancy. As described, the syndrome comprised changes in the epicanthic folds, a flat nasal bridge, a small upturned nose, a long upper lip with a relatively shallow philtrum, a thin upper vermillion border and downturned angles of the mouth. In two of their seven foetuses, hypospadias and delay in psychomotor development were present.
Over the subsequent years, this initial report was followed by numerous further accounts of the occurrence of this particular syndrome, some of the accounts attempting to add further features to the clinical picture. By 2001 Kozma (2001) had collected 69 instances from the literature and added two further examples. This writer at that time described the features of the syndrome as involving a consistent facial phenotype characterized by a small broad nose, small ears, flat philtrum, long upper lip with shallow philtrum and micrognathia or retrognathia. It was associated with multiple systemic and orthopaedic abnormalities (in 62 %), central nervous system dysfunction and altered physical growth. Minor skin defects were present in 30 %, cardiovascular abnormalities in 26 %, genital abnormalities in 22 % and lung abnormalities in 16 %. Brain, eye, kidney and hearing defects were less frequent, while neural tube defects occurred in 3 %. There was a substantial death rate during infancy in those affected (12 %), while another 23 % exhibited developmental defects or mental retardation.
This constellation of abnormalities did not limit the spectrum of abnormal developmental manifestations which later authors attempted to include in the syndrome, for instance, speech delay, joint laxity, glue ears and autistic features (Moore et al. 2000), the rare so-called Baller–Gerold syndrome that features malformation of the skull, face, forearm and hand bones (Lype et al. 2008) and septo-optic dysplasia (McMahon and Braddock 2001). Jacobsen et al. (2014) reported an increased risk of dental agenesis associated with valproate exposure in utero.
In the meanwhile, reports of larger-scale studies of the consequences of intrauterine valproate exposure had begun to appear (e.g. Koch et al. 1992; Kaaja et al. 2003; Artama et al. 2005; Wyszynski et al. 2005). These were among the 10 publications that Wlodarczyk et al. (2012) considered in their paper, in which they reported that the major congenital malformation rates associated with valproate exposure had ranged from 5.7 to 16.8 %. In the seven studies for which odds ratios or relative risks had been calculated, the values ranged between 2.52 and 5.94, in all instances being statistically significant at the P < 0.05 level. Miki et al. (2014) subsequently published a meta-analysis of 58 cohort studies relevant to valproate-associated teratogenesis in humans, including some of those discussed immediately above.
In an analysis of the Australian Pregnancy Register data accumulated by late 2012, foetal malformations had occurred in 13.8 % of 253 pregnancies in which valproate had been used in monotherapy, a relative risk of 4.23 (95 % C.I. = 1.69, 10.57) compared with that for malformations in women with epilepsy who did not take antiepileptic drugs during at least the earlier months of pregnancy.
There thus seems quite strong evidence that valproate is a significant human teratogen . Further, a number of studies have shown that the malformation rate associated with the drug is dose dependent. Omtzigt et al. (1992) had noted that the occurrence of spina bifida was associated with statistically significantly higher valproate doses (mean 1640 ± 136 compared with 941 ± 48 mg per day). Canger et al. (1999) reached a similar conclusion. Samrén et al. (1999) reported that the malformation rate associated with valproate doses above 1000 mg per day was significantly higher than the rate for lower doses. Kaneko et al. (1999) plotted malformation rate against valproate dose and showed the malformation risk increased with drug dose. Vajda et al. (2004) and Vajda and Eadie (2005) observed in the data of the Australian Pregnancy Register that the valproate-associated malformation rate was substantially higher at daily drug doses in excess of 1400 mg a day during pregnancy and initially suggested that this value might distinguish between comparatively safe and unacceptably hazardous dosages from the foetal standpoint. Shortly afterwards, Vajda et al. (2006) reduced the cut-off dose to 1100 mg per day. Diav-Citrin et al. (2008) nominated a dose of 1000 mg a day as separating a 21.9 % malformation risk from a 2.5 % one. Later analyses of expanded data from the Australian Register , employing logistic regression, have shown that in practice there probably is no really safe valproate dosage from the standpoint of avoiding foetal malformation (Eadie 2008 and Figs. 8.1, 8.2 and 8.3).
The existence of this dose dependence in malformation rate associated with valproate has been confirmed in further studies (Bromfield et al. 2008; Tomson et al. 2011) and seems to be to be fairly generally accepted in the recent literature. The combination of the greater tendency of valproate to be associated with foetal malformations and the dose relatedness of the risk may explain much of the rather wide range of published values for the overall hazard of foetal malformation associated with intrauterine antiepileptic drug exposure in general. The more pregnancies exposed to valproate in a series of antiepileptic drug-treated pregnancies, the higher the malformation rate in that series will appear to be. Even in two series with equal proportions of valproate-exposed pregnancies, the overall malformation rate will be higher in the series with the greater mean valproate dose. As mentioned earlier (Chap. 8), the combination of the two factors also seems to explain the rather frequently described earlier finding that use of antiepileptic drugs in polytherapy was associated with higher malformation rates than use of the drugs in monotherapy.
Vajda et al. (2013b) have noted another phenomenon in relation to valproate dosage. At doses above 2000 mg a day, exposure to the drug tends to be statistically significantly associated with the occurrence of spina bifida and hypospadias and at lower doses with other malformations (Fig. 9.1). This suggests that the type of malformation associated with exposure to an antiepileptic drug may depend on the drug dose involved. This finding obviously needs confirmation, though that may be difficult to obtain because the already existing data about the dose dependence of the malformation risk associated with the drug has led to valproate being used in lower dosages, if it must be used in pregnant women. This use of lower dosages of valproate has been associated with a parallel decline in foetal malformation rates in the Australian data (Vajda et al. 2013b; Fig. 9.2).
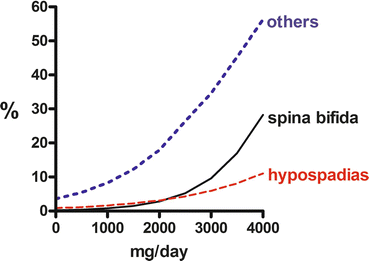
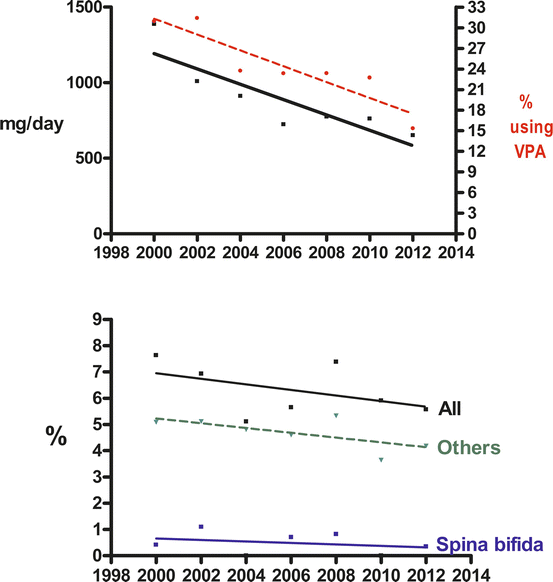
Fig. 9.1
Logistic regressions for risk of spina bifida (continuous line), hypospadias (lower broken line) and for all malformations except spina bifida and hypospadias (upper dotted line) on valproate dose. All regressions are statistically significant
Fig. 9.2
Top panel: linear regressions on date for (i) mean valproate dose (continuous line – dose values shown on left ‘y’ axis) prescribed during consecutive 2-year periods between 1999 and 2014 and for (ii) the proportion of all pregnancies in the Australian Register that were prescribed valproate (percentage values shown on right ‘y’ axis; regression, broken line). Lower panel: linear regressions on dates for percentages of valproate exposed pregnancies in which (i) all types of malformation (upper continuous line) (ii) spina bifida only (lower continuous line) and (iii) all foetal malformations apart from spina bifida (dashed line) occurred. The slopes of all the linear regressions in both panels, except that for all other malformations, are statistically significant
Lamotrigine
Lamotrigine was first marketed in 1991, though its introduction into the United States was further delayed. The drug is increasingly being employed in the management of pregnant women with epilepsy and currently enjoys a reputation for being among the safest of the antiepileptic drugs from the teratogenicity point of view. Sabers et al. (2004) reported a 2.0 % malformation rate associated with foetal exposure to the drug. Morrow et al. (2006) analysed the data of the United Kingdom Register and found a major congenital malformation rate of 3.2 % (95 % C.I. = 2.1, 4.9 %) in 647 pregnancies in which the drug had been used in monotherapy. The malformation rate for lamotrigine was nonstatistically significantly higher than the 2.2 % rate for carbamazepine , the major comparator used (odds ratio 1.71; 95 % C.I. = 0.88, 3.32). Morrow et al. also noted that the frequency of malformations appeared to increase progressively through the lamotrigine dosage bands of (i) up to 100 mg per day, (ii) 100–200 mg per day and (iii) over 200 mg per day. The mean lamotrigine dose associated with malformations, 352.4 mg per day, was higher than the mean dose in pregnancies where there was no malformation (250.6 mg per day, P < 0.0001). At much the same time, Vajda et al. (2006) described 65 pregnancies in which lamotrigine was used as the only epileptic drug and in which no foetal malformations had occurred, and Meador et al. (2006), by means of a literature review, traced 98 pregnancies involving lamotrigine monotherapy in which there was a serious outcome, including major congenital malformations , in only 1 %.
Cunnington et al. (2007) reported that there had been a major congenital malformation rate of 2.7 % (95 % C.I. = 1.8, 4.2 %) for 802 pregnancies in the records of the firm marketing the drug (this population probably included some or all of the pregnancies in the studies mentioned immediately above). Regression analysis found no relationship between the risk of malformations and the drug dose. In the North American Registry data, Holmes et al. (2008) calculated that there was a 2.3 % major congenital malformation rate for the drug. Compared with 206,224 antiepileptic drug-unexposed infants in whom there was a cleft palate or cleft lip rate of 0.07 %, they found a 10.4-fold increase in the incidence of cleft palates or lips in the pregnancies exposed to lamotrigine in monotherapy. Then Mawer et al. (2010) described a 5.4 % major congenital malformation rate in 227 pregnancies in lamotrigine-treated women with epilepsy, as compared with a 2.1 % rate in 315 matched control pregnancies (P = 0.23). Vajda et al. (2010) found a 4.9 % malformation rate in 243 pregnancies exposed to lamotrigine monotherapy, as compared with a 3.4 % malformation rate in 118 pregnancies in women with epilepsy that was untreated during the critical period of organogenesis (odds ratio 1.48; 95 % C.I. = 0.46, 4.69). Unlike the situation for valproate , logistic regression for malformation risk plotted against increasing drug dose had a downward slope in the Vajda et al. data. Molgaard-Nielsen and Hviid (2011) linked a Danish database of 837,795 live births between 1996 and 2008 with a database for national dispensed antiepileptic drugs. Major congenital malformations had occurred in 3.7 % of 1019 lamotrigine-exposed pregnancies, an adjusted prevalence odds ratio of 1.18 (95 % C.I. = 0.83, 1.68) compared with that which applied for the general population. In the same year Tomson et al. (2011), based on the EURAP data, quoted a major foetal malformation rate of 2.0 % for lamotrigine monotherapy in doses below 300 mg a day, based on the set of 1280 pregnancies in which the drug had been used in monotherapy. These authors stated that the malformation rate associated with lamotrigine was dose dependent. Holmes and Hernandez-Diaz (2012) described a 2 % foetal malformation rate in 1562 pregnancies exposed to lamotrigine in monotherapy (relative risk versus controls 1.8; 95 % C.I. = 0.7, 4.6).
Then Campbell et al. (2014) provided updated data from the United Kingdom Pregnancy Register at a stage when it contained 2198 pregnancies in which lamotrigine had been used as the sole antiepileptic drug. In these pregnancies there was a major congenital malformation rate of 2.3 % (95 % C.I. = 1.8, 3.1 %), and unlike the earlier analysis of this register ’s contents, the trend for the risk of malformation to increase with increasing drug dose was not statistically significant. Veiby et al. (2014), based on 833 Norwegian pregnancies in which lamotrigine was used in monotherapy, calculated a 3.4 % malformation rate with an odds ratio of 1.26 (95 % C.I. = 0.87, 1.84) compared with a control group of 771,412 antiepileptic drug-unexposed children. Vajda et al. (2014) studied data from the Australian Pregnancy Register at the end of 2013. There was a major congenital malformation rate of 4.6 % in the 307 lamotrigine monotherapy-exposed pregnancies, with a relative risk of malformation of 1.40 (0.51, 3.80) compared with 154 untreated pregnancies in women with epilepsy. Logistic regression analysis showed that the resulting regression line had a slight and nonstatistically significant downward slope for malformation risk on increasing drug dose (Fig. 8.2).
Thus there is a reasonable amount of relatively recent data concerning the foetal malformation hazard associated with use of lamotrigine in the pregnancies of women with epilepsy, though some of the material has probably been included more than once in the analysed data sets. Consistently, the malformation rates have been a little higher than those for the comparator populations that different groups of workers have chosen. However, none of the rates has been statistically significantly higher. Some studies have suggested that the foetal hazards from the drug may be dose related, though the evidence for this never achieved the P < 0.05 level of statistical significance. Studies employing logistic regression have not suggested that such a dosage dependency exists. Interestingly, in the Australian data where regression analysis provided no evidence for a dose-dependent risk, when the malformation rate was correlated with the three lamotrigine dosage bands that Morrow et al. (2006) used, the malformation rate appeared to increase with dosage. However, if the highest dosage band was subdivided into two, the rate was lower for the new highest dosage range than for the immediately lower one. It seems reasonable to conclude that, if lamotrigine is a teratogen , its teratogenic potential is comparatively small.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


