32 Parkinson’s disease
Aetiology
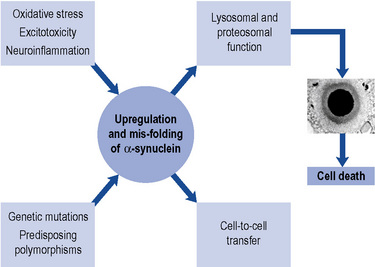
In a small number of patients, genetic factors are dominant. The discovery of a mutation in the gene coding for a synaptic protein called α-synuclein has provided tremendous impetus for further research. Such mutations have been described in fewer than 10 families worldwide. Nevertheless, because α-synuclein is a major component of the pathological hallmark of Parkinson’s disease, the Lewy body (see below), the challenge is to discover how a mutation in this protein in a tiny minority can relate to the formation of Lewy bodies in the vast majority. In recent years, eight genetic loci and a further four genes (parkin, DJ-1, PINK1 and LRRK-2) have been identified (Healy et al., 2004). The intriguing thing is that the protein products of these genes are involved in a cellular system called the ubiquitin-proteasome system, which plays a crucial role in removing and recycling abnormal or damaged proteins. Current thinking is that abnormalities in the way in which the cell handles mutated or abnormal proteins may ultimately lead to its death, through increased oxidative stress and/or reduced mitochondrial energy production. The Lewy body may actually represent a defence mechanism by the cell to ‘parcel up’ potentially damaging proteinaceous material (Olanow et al., 2004).
More recently, cell-to-cell transfer of α-synuclein has been demonstrated in vitro and also in engrafted stem cell tissue. This suggests that the pathology of Parkinson’s disease may be propagated between neurones and could have major implications for the spread of Lewy body pathology within the brain, as well as its treatment (Olanow and Prusiner, 2009) (Fig. 32.1).
Pathophysiology
The characteristic pathological features of Parkinson’s disease are neuronal loss in pigmented brainstem nuclei, together with the presence of eosinophilic inclusion bodies, called Lewy bodies, in surviving cells. The pars compacta of the substantia nigra in the midbrain is particularly affected. Dopaminergic neurones within this nucleus project to the striatum, which is, therefore, deprived of the neurotransmitter dopamine. In Parkinson’s disease, there is a loss of over 80% of nigral neurones before symptoms appear. The ‘Braak hypothesis’ has been proposed to account for spread of pathology within the Parkinsonian brain and suggests that α-synuclein may first accumulate in the lower brainstem and then gradually ascend rostrally to affect critical brain regions including the substantia nigra and ultimately the cerebral cortex (Braak et al., 2003).
Differential diagnosis
It is important to remember that, while Parkinson’s disease is a common form of Parkinsonism, there are numerous other degenerative and symptomatic causes. Further, ‘all that shakes is not Parkinson’s disease’. Table 32.1 gives a differential diagnosis for causes of Parkinsonism. These are separated into degenerative and symptomatic categories. The list is not exhaustive and excludes, for instance, rare Parkinsonian manifestations in uncommon diseases. A detailed description of these different causes of Parkinsonism is beyond the scope of this chapter, but a few points should be highlighted. Essential tremor is not included in Table 32.1, as this common condition does not cause bradykinesia. Nevertheless, it may be very difficult to differentiate from tremor-dominant Parkinson’s disease. A positive family history and good response to alcohol may provide vital clues towards the diagnosis of essential tremor, although in practice these are not always reliable.
Table 32.1 Differential diagnosis of Parkinsonism
| Degenerative causes | Symptomatic causes |
|---|---|
| Parkinson’s disease | Dopamine receptor blocking agents |
Several clinical and clinicopathological series have confirmed our fallibility in not making a correct diagnosis of Parkinson’s disease. If clinical criteria, such as those produced by the UK Parkinson’s Disease Brain Bank, are not applied, then the error rate (false-negative diagnosis) may be as high as 25–30%. These criteria are listed in Box 32.1. Degenerative conditions commonly masquerading as Parkinson’s disease include progressive supranuclear palsy, multiple system atrophy and Alzheimer’s disease.
Box 32.1 Clinical criteria for diagnosis of Parkinson’s disease
Step 1 Diagnosis of Parkinsonian syndrome
The patient has bradykinesia, plus one or more of the following:
Drug-Induced Parkinsonism
Perhaps the most important differential diagnosis to consider when a patient presents with Parkinsonism is whether their symptoms and signs may be drug induced. This is because drug-induced Parkinsonism is potentially reversible upon cessation of the offending agent. Reports linking drug-induced Parkinsonism with the neuroleptic chlorpromazine were first published in the 1950s. Since then, numerous other agents have been associated with drug-induced Parkinsonism. Many of these are widely recognised, although others are not (Box 32.2). Compound antidepressants were a problem in the past because they contained neuroleptic drugs. For example, fluphenazine was found with nortriptyline in Motival® (discontinued in the UK in 2006) and often overlooked as a potential culprit. Repeat prescription of vestibular sedatives and anti-emetics such as prochlorperazine and cinnarizine are other commonly encountered causes of drug-induced Parkinsonism. The pathogenesis of drug-induced Parkinsonism is unlikely to be only due to dopamine receptor blockade. If this were the case, the incidence and severity should correlate with the drug dosage and length of exposure, and this is not clearly observed. Sodium valproate is also now recognised to cause an encephalopathy dominated by Parkinsonism and cognitive impairment which is reversible upon drug cessation. Again, there is considerable idiosyncrasy in who develops this encephalopathy when exposed to valproate.
Box 32.2 Examples of non-neuroleptic drugs associated with drug-induced Parkinsonism
a Lithium causes postural tremor. Reports of Parkinsonism occurring with lithium have usually been in the context of prior exposure to neuroleptics.
b Only single case reports of drug-induced Parkinsonism with these drugs.
c One case report of drug-induced Parkinsonism in a child after bone marrow transplantation and a second in association with cytosine arabinoside therapy.
Investigations
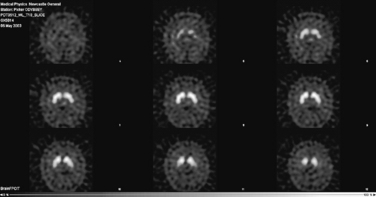
The diagnosis of Parkinson’s disease is a clinical one and should be based, preferably, upon validated criteria. In young-onset or clinically atypical Parkinson’s disease, a number of investigations may be appropriate. These include copper studies and DNA testing to exclude Wilson’s disease and Huntington’s disease, respectively. Brain imaging by computed tomography (CT) or magnetic resonance imaging (MRI) may be necessary to exclude hydrocephalus, cerebrovascular disease or basal ganglia abnormalities suggestive of an underlying metabolic cause. When there is difficulty in distinguishing Parkinson’s disease from essential tremor, a form of functional imaging called FP-CIT SPECT (also known as DaTSCAN) may be useful, as this technique can sensitively identify loss of nigrostriatal dopaminergic terminals in the striatum (Fig. 32.2). Thus, in essential tremor, the SPECT scan is normal, whereas in Parkinson’s disease, reduced tracer uptake is seen (Jennings et al., 2004).
Treatment
General approach
When treatment becomes necessary, it is impossible to generalise about which drug should be commenced. All currently available drugs for Parkinson’s disease are symptomatic, as no agent has yet been shown, beyond reasonable doubt, to have disease-modifying or neuroprotective properties. There is no accepted algorithm for the treatment of Parkinson’s disease, although a clinical management guideline has been produced (NICE, 2006).
A number of factors, including patient preference, age, severity and type of disease (tremor-dominant versus bradykinesia-dominant) and co-morbidity, need to be taken into account. The efficacy and tolerability of levodopa in Parkinson’s disease was first described 1967, when the drug was started in low doses and gradually increased thereafter (Cotzias et al., 1967).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


