Pancreatitis
KEY CONCEPTS
ACUTE PANCREATITIS
![]() Factors that can contribute to acute pancreatitis should be corrected, including discontinuation of medications that could be potential causes.
Factors that can contribute to acute pancreatitis should be corrected, including discontinuation of medications that could be potential causes.
![]() Patients with acute pancreatitis without the systemic inflammatory response syndrome should receive aggressive fluid replacement, but goal-directed therapy has not been defined.
Patients with acute pancreatitis without the systemic inflammatory response syndrome should receive aggressive fluid replacement, but goal-directed therapy has not been defined.
![]() Patients with severe acute pancreatitis and the systemic inflammatory response syndrome require early and aggressive IV fluid resuscitation and should be managed similarly to patients with sepsis.
Patients with severe acute pancreatitis and the systemic inflammatory response syndrome require early and aggressive IV fluid resuscitation and should be managed similarly to patients with sepsis.
![]() Parenteral opioid analgesics are used to control abdominal pain associated with acute pancreatitis.
Parenteral opioid analgesics are used to control abdominal pain associated with acute pancreatitis.
![]() The only definitive indication for antibiotic use in acute pancreatitis is to treat known or suspected infection.
The only definitive indication for antibiotic use in acute pancreatitis is to treat known or suspected infection.
CHRONIC PANCREATITIS
![]() Chronic pain, malabsorption with resultant steatorrhea, and diabetes mellitus are the hallmark complications and symptoms of chronic pancreatitis.
Chronic pain, malabsorption with resultant steatorrhea, and diabetes mellitus are the hallmark complications and symptoms of chronic pancreatitis.
![]() Pain from chronic pancreatitis can initially be treated with nonopioid analgesics, but opioids will eventually be required as the disease progresses.
Pain from chronic pancreatitis can initially be treated with nonopioid analgesics, but opioids will eventually be required as the disease progresses.
![]() Reduction in dietary fat intake and pancreatic enzyme supplementation are the primary treatments for malabsorption due to chronic pancreatitis.
Reduction in dietary fat intake and pancreatic enzyme supplementation are the primary treatments for malabsorption due to chronic pancreatitis.
![]() Enteric-coated pancreatic enzyme supplements are the preferred dosage form in the treatment of malabsorption and steatorrhea due to chronic pancreatitis.
Enteric-coated pancreatic enzyme supplements are the preferred dosage form in the treatment of malabsorption and steatorrhea due to chronic pancreatitis.
![]() The addition of an antisecretory agent to pancreatic enzyme supplementation may increase the effectiveness of enzyme therapy for malabsorption and steatorrhea due to chronic pancreatitis.
The addition of an antisecretory agent to pancreatic enzyme supplementation may increase the effectiveness of enzyme therapy for malabsorption and steatorrhea due to chronic pancreatitis.
Pancreatitis is inflammation of the pancreas with variable involvement of regional tissues or remote organ systems.1,2 Acute pancreatitis is characterized by severe pain in the upper abdomen and elevations of pancreatic enzymes in the blood.2 In the majority of patients, acute pancreatitis is a mild, self-limiting disease that resolves spontaneously without complications. Approximately 20% of adults with acute pancreatitis have a severe course, and 10% to 30% of those with severe acute pancreatitis die.1,2 Severe pancreatitis with either organ failure or infected necrosis is associated with a mortality of approximately 30% and it increases when both are present.3 Although exocrine and endocrine pancreatic functions may remain impaired for variable periods after an attack, acute pancreatitis usually does not progress to chronic pancreatitis.4
Chronic pancreatitis is characterized by long-standing inflammation that eventually leads to a loss of pancreatic exocrine and endocrine functions.4–6 It is a progressive disease that often goes unnoticed for many years. Usually patients first present with complaints of chronic abdominal pain. Later in the disease process malabsorption with resultant steatorrhea occurs. This leads to malnutrition and weight loss. Finally, patients develop diabetes mellitus due to a loss of pancreatic endocrine function.4,5
EPIDEMIOLOGY
The prevalence of pancreatitis varies widely with geographic, etiologic (e.g., alcohol consumption), environmental, and genetic factors. Hospitalizations for acute pancreatitis have increased in the United States, most likely related to an increase in gallstones in association with obesity.7 Admission rates for acute pancreatitis are approximately 40 per 100,000 per year in the United States.7 Approximately 6 per 100,000 population will develop chronic pancreatitis with a peak incidence between ages 35 and 54 and about 85% of cases occurring in men.6 However, this incidence may be underestimated due to diagnostic difficulties and various classification systems. Also, the prevalence of chronic pancreatitis varies widely based on geographic location.4,6 Hospitalization for chronic pancreatitis has also doubled in the past decade with black patients being almost two to three times as likely to be hospitalized for chronic pancreatitis than for alcoholic cirrhosis.6
PANCREATIC EXOCRINE PHYSIOLOGY
The pancreas possesses both endocrine and exocrine functions. The islets of Langerhans, which contain the cells of the endocrine pancreas, secrete insulin, glucagon, somatostatin, and other polypeptide hormones. The exocrine pancreas is composed of acini and ductules that secrete about 2.5 L/day of isotonic fluid that contains water, electrolytes, and pancreatic enzymes necessary for digestion. Bicarbonate and other electrolytes are secreted primarily by the centroacinar (ductular) cells in order to neutralize gastric acid. Pancreatic juice is delivered to the duodenum via the pancreatic ducts (Fig. 25-1) where the alkaline secretion neutralizes gastric acid and provides an appropriate pH for maintaining the activity of pancreatic enzymes.8
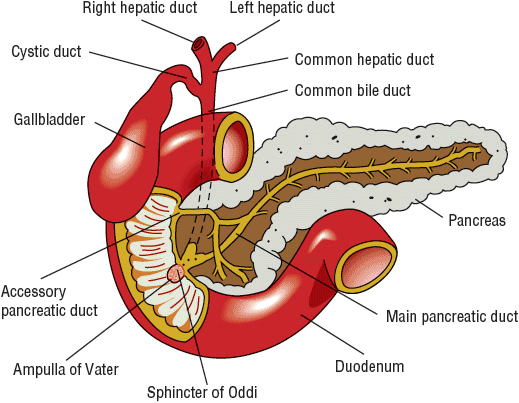
FIGURE 25-1 Anatomic structure of the pancreas and biliary tract.
The major pancreatic exocrine enzyme groups are as follows:
1. Amylolytic: amylase
2. Lipolytic: lipase, procolipase, prophospholipase A2, and carboxylesterase
3. Proteolytic: trypsinogen, chymotrypsinogen, procarboxy-peptidase, and proelastase
4. Nucleolytic: ribonuclease and deoxyribonuclease
5. Other: trypsin inhibitor
Amylase is responsible for digestion of starches and glycogen through hydrolysis. The lipolytic enzymes break down triglycerides, cholesterol, and other fats in the digestive tract. Specifically, lipase hydrolyzes triglycerides into fatty acids and monoglycerides. Colipase and bile acids facilitate this process by allowing lipase to act on the hydrophobic surface of fat droplets in the mainly hydrophilic environment. Phospholipase A2 and carboxylesterase continue to break down fatty acids, cholesterol, monoglycerides, and other products of fat digestion. Proteolytic enzymes digest proteins into oligopeptides and free amino acids, while nucleases break down nucleic acids.8,9
The production of proteolytic enzymes in the pancreas occurs in a manner that prevents self-digestion of the pancreas. These enzymes are synthesized within the acinar cells and secreted into the duodenum as zymogens (inactive enzymes). Enterokinase secreted by the duodenal mucosa converts trypsinogen to trypsin, which then activates all other proteolytic zymogens along with procolipase and prophospholipase A2. Thus, two important mechanisms protect the pancreas from the potential degradative action of its own digestive enzymes. First, the synthesis of proteolytic enzymes as zymogens requires extrapancreatic activation by trypsin. Second, pancreatic juice contains a low concentration of trypsin inhibitor, which inactivates any autocatalytically formed trypsin within the pancreas. Proteolytic activity of trypsin in the intestinal lumen is not inhibited because the concentration of trypsin inhibitor is minimal. Lipase, amylase, ribonuclease, and deoxyribonuclease are secreted by the acinar cells in their active form.8
The regulation of exocrine pancreatic secretion is a complex interplay of neurohormonal feedback with three distinct phases. The first phase is the cephalic phase where the sight, smell, and taste of food produce pancreatic enzyme secretion through stimulus of the vagus nerve. Vasoactive intestinal peptide (VIP) and gastrin-releasing peptide (GRP) released from efferent vagus nerve terminals bind to receptors on the acinar cells stimulating enzyme release.8 Water and bicarbonate are also released from ductal cells due to VIP stimulation. The gastric phase occurs due to gastric distension from food entering the stomach. This results primarily in secretion of digestive enzymes from the pancreas. Once chyme enters the duodenum, the intestinal phase begins. The chyme causes secretin to be released from the duodenal mucosa when its pH is less than 4.5. Secretin results in water and bicarbonate secretion from the pancreas, which is necessary since lipolytic enzymes are inactivated at a pH below 5.9 Digestive enzymes are released from the pancreas due to the presence of fatty acids, peptides, amino acids, and glucose in the duodenum.8
The feedback mechanism for continued release of pancreatic enzymes involves the hormone cholecystokinin (CCK). When products of fat, protein, and starch digestion enter the upper small intestine, they stimulate release of CCK from I cells into the blood. Elevated levels of CCK in the serum activate a vagovagal reflex causing further release of VIP and GRP, leading to enhanced pancreatic enzyme secretion. Inhibition of this feedback loop is thought to be due to trypsin. After digestion is complete, unoccupied trypsin is thought to inhibit the release of CCK.8 A more in-depth discussion of pancreatic physiology can be found elsewhere.8
ACUTE PANCREATITIS
Acute pancreatitis varies from mild to severe disease. The morphologic appearance of the pancreas and surrounding tissue ranges from interstitial edema and inflammatory cells (interstitial pancreatitis) to pancreatic and extrapancreatic necrosis (necrotizing pancreatitis). Necrotizing pancreatitis has a higher risk of infection, organ failure, and mortality.10,11 The rupture of blood vessels within or around the pancreas can also lead to a collection of blood in the retroperitoneal space.
Etiology
Table 25-1 lists the etiologic risk factors associated with acute pancreatitis. Obstruction caused by gallstones is the most common cause of acute pancreatitis and alcohol abuse the second; together they account for 70% to 80% of all cases of acute pancreatitis.6 Genetic and autoimmune causes of acute pancreatitis have been identified.14 Most of the remaining cases are idiopathic.2 Acute pancreatitis can occur as a result of an endoscopic retrograde cholangiopancreatography (ERCP) procedure and is more common following therapeutic ERCP than diagnostic, with overall rates of 1.6% to 5.4% and 0.4% to 5.4%, respectively. High-risk populations may have rates of acute pancreatitis as high as 15%.15 Cigarette smoking appears to increase the risk of pancreatitis, especially in alcohol-related disease.16 Pregnancy is not considered a cause of acute pancreatitis; however, pregnant women develop pancreatitis as a result of a coincident process, most commonly cholelithiasis. In pediatric patients, the common etiologies are systemic illness, biliary disease, trauma, and medications.17
TABLE 25-1 Etiologic Risk Factors Associated with Acute Pancreatitis

Medications
Drug-induced acute pancreatitis should be suspected when other causes have been excluded and there is a temporal relationship with the initiation of a medication that has been implicated as a cause. The percentage of acute pancreatitis cases caused by medications is reported to be 0.1 to 2.18 Most information on drug-induced acute pancreatitis is obtained from case reports, which do not provide reliable information on incidence. The most convincing case reports involve recurrence on rechallenge; however, rechallenge is rare, occurring only when alternative therapy is not available. When evaluating case reports, clinicians should review data supporting the diagnosis of acute pancreatitis, attempts to rule out other causes, and the onset of acute pancreatitis in relation to drug therapy. Further complicating the evaluation of some reports is that certain patient populations may have an increased risk of pancreatitis.
Proton pump inhibitors and histamine2-receptor antagonists may be initiated in response to early symptoms of unrecognized pancreatitis and may confound the association between the drug and the disease. However, a retrospective cohort study does not support an association between acute pancreatitis and proton pump inhibitors or histamine2-receptor antagonists.19 Medications that alter serum lipid concentrations, such as propofol and tamoxifen, are associated with pancreatitis from hyperlipidemia.20,21 In contrast, a meta-analysis of lipid-lowering therapies found that statins were associated with a decreased number of acute pancreatitis cases.22 Pancreatitis was not a stated end point of any of the trials included and there are numerous case reports of apparent drug-induced pancreatitis with statins. There is a higher incidence of drug-induced acute pancreatitis in U.S. patients with human immunodeficiency virus (HIV) treated with antiretroviral therapy.23 However, there was no increase in acute pancreatitis associated with antiretroviral use in a well-controlled trial including data from 33,742 person-years.24 Medications used in the treatment of inflammatory bowel disease and type II diabetes mellitus have also been associated with a higher incidence of drug-induced acute pancreatitis.25,26 In addition, patients with diabetes mellitus may also have an inherent increased risk of acute pancreatitis. The commonly prescribed medication metformin is associated with acute pancreatitis at toxic serum concentrations.27 There have also been case reports of acute pancreatitis in patients receiving some of the newer antihyperglycemic agents, including the dipeptidyl peptidase-4 (DPP-4) inhibitors, sitagliptin, linagliptin, and vildagliptin, and the glucagon-like peptide-1 (GLP-1) receptor agonists, exenatide and liraglutide.18 Exenatide and liraglutide continue to account for a substantial percentage of drug-induced pancreatitis reported to the U.S. FDA.28
The onset of drug-induced pancreatitis after initiation of medications ranges from a few months to several years, with a median of 5 weeks; onset after rechallenge can occur within hours. The onset may differ according to the mechanism. Clinicians should be especially suspicious of drug-induced acute pancreatitis in high-risk patients, such as those receiving immunomodulating drugs or who have HIV infection, the elderly, or those with diabetes mellitus.18
Mechanisms of drug-induced pancreatitis are not clearly defined but may fall into several general categories, including direct toxic effects of the drug or its metabolites, hypersensitivity, drug-induced hypertriglyceridemia, and alterations of cellular function in the pancreas and pancreatic duct.26 Once the process is initiated, disease severity is determined by the propagation of proinflammatory mediators. Although acute pancreatitis is an infrequent complication of drug therapy, it is prudent to withdraw medication when an association is suspected.
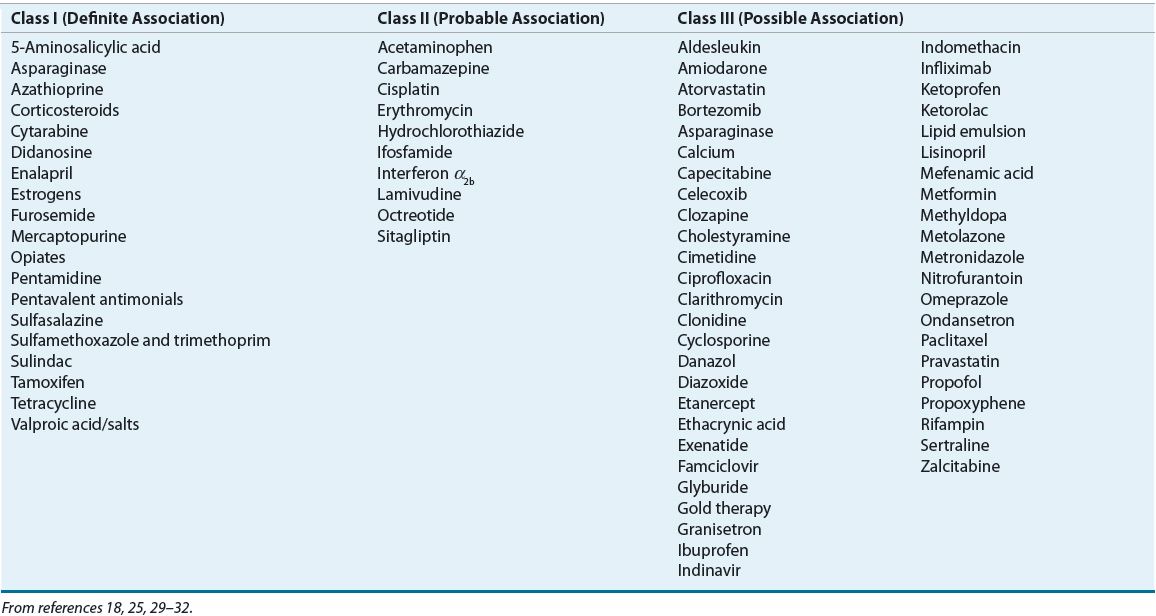
Numerous drugs are believed to cause acute pancreatitis, but ethical and practical considerations prevent rechallenge with suspected agents. Table 25-2 lists specific agents associated with acute pancreatitis based on known association. Class I (definite association) implies a temporal relationship of drug administration to abdominal pain and hyperamylasemia in at least 20 reported cases with at least one positive response to rechallenge with the offending agent. Class II medications are implicated in more than 10, but less than 20, reported cases of acute pancreatitis and suggest a probable association. Class III includes medications with a possible association, defined as fewer than 10 published cases or unpublished reports in pharmaceutical or FDA files. Table 25-2 only includes selected class III medications. A comprehensive list of class III drugs can be found elsewhere.33
TABLE 25-2 Medications Associated with Acute Pancreatitis
Pathophysiology
The pathophysiology of acute pancreatitis is based on events that initiate injury and secondary events that establish and perpetuate the injury (Fig. 25-2). Alcohol abuse and gallstones cause different initial insults to the pancreas. However, acute pancreatitis of any etiology has long been thought to result from the premature activation of trypsinogen to trypsin within the pancreas, leading to activation of other digestive enzymes and autodigestion of the gland.2 The lysosomal proteinase, cathepsin B, and intracellular calcium may be involved in the activation of trypsinogen as well as decreased activity of trypsin inhibitor.34 Genetic abnormalities in pathways that protect the pancreas from autodigestion also play a pathophysiologic role in the development of some forms of acute pancreatitis, and may be the differentiating factor in the minority of alcoholics who develop disease.35
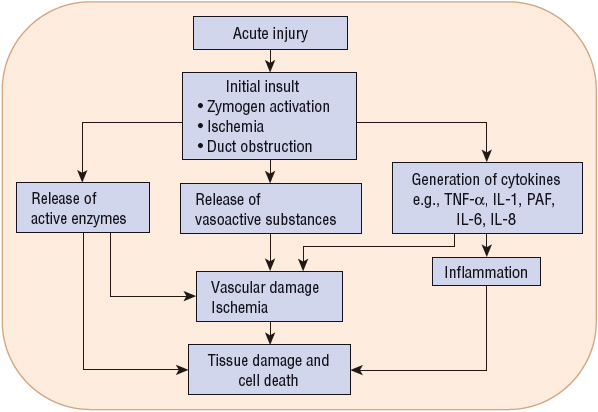
FIGURE 25-2 Pathophysiology of acute pancreatitis: initiating and secondary events. (IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; PAF, platelet-activating factor; TNF-α, tumor necrosis factor-α.)
In addition to increased production, activated enzymes are retained in the acinar cells in higher concentrations than normal.36 Activated pancreatic enzymes released into the pancreas and surrounding tissues produce damage and necrosis to the pancreatic tissue, the surrounding fat, the vascular endothelium, and adjacent structures. Lipase damages the fat cells, producing noxious substances that cause further pancreatic and peripancreatic injury. The release of cytokines by acinar cells directly causes their injury and enhances the inflammatory response.34 Injured acinar cells liberate chemoattractants that recruit neutrophils, macrophages, and other cells to the area of inflammation. These immune responses cause a systemic inflammatory response syndrome (SIRS). Vascular damage and ischemia causes the release of kinins, which makes capillary walls permeable and promotes tissue edema. The release of damaging oxygen-free radicals appears to correlate with the severity of pancreatic injury.10 Finally, pancreatic infection may result from increased intestinal permeability and translocation of colonic bacteria.35
Complications
Early complications are a result of fluid losses and SIRS. Hypotension results from hypovolemia, hypoalbuminemia, the release of kinins, and sepsis. Even patients with mild disease have significant fluid losses. Renal complications are usually caused by hypovolemia. The most common systemic complication of acute pancreatitis is respiratory failure.1 GI bleeding occurs secondary to numerous causes including rupture of a pseudocyst. Severe acute pancreatitis is also associated with confusion and coma.
Local complications—including acute fluid collection, pancreatic necrosis, infection, abscess (collection of pus in or adjacent to the pancreas), and pseudocyst—develop approximately 3 to 4 weeks after the initial attack. Pancreatic infections occur in 15% to 30% of those with pancreatic necrosis and are usually secondary infections of necrotic tissue.7 Pancreatic ascites occurs when pancreatic secretions spread throughout the peritoneal cavity. Systemic complications include cardiovascular, renal, pulmonary, metabolic, hemorrhagic, and CNS abnormalities.10 Long-term complications include glucose intolerance and recurrence of acute pancreatitis.37
Clinical Presentation
Signs and Symptoms
The clinical presentation of acute pancreatitis varies depending on the severity of the inflammatory process and whether damage is confined to the pancreas or involves local and systemic complications (Table 25-3).7
TABLE 25-3 Presentation and Diagnosis of Acute Pancreatitis

Diagnosis
Most guidelines agree that the diagnosis should be made within 48 hours based on characteristic abdominal pain and amylase, lipase, or both that are elevated to at least three times the upper limit of normal. Lipase is more sensitive and specific than amylase and is preferred. Contrast-enhanced computed tomography (CECT) of the abdomen may be used to confirm the diagnosis, including in patients with amylase or lipase that is not three times the upper limit of normal. Some guidelines consider ultrasonography to be an acceptable alternative to CECT.1,10,41 However, it is best used to ascertain the presence of gallstones. The diagnosis of acute pancreatitis should also be considered when evaluating patients with SIRS (see Table 25-3).10,41,42 For further information on laboratory tests and abdominal imaging, refer to Table 25-3.
Prediction of Disease Severity Prediction of severity of acute pancreatitis is useful for decisions involving the need for aggressive treatment, including admission to an intensive care unit. The risk for severe acute pancreatitis should be assessed on admission and on an ongoing basis.10,11 Several scoring systems have been developed to assess the likelihood of severe disease (Table 25-4). However, development and validation of such systems remains an ongoing area of research. Scoring systems are developed based on retrospectively identified associations between clinical and laboratory findings and morbidity and mortality.1,43,44 Many are too complicated for bedside use or rely on measurements that are not widely available. Some scoring systems have not been validated in prospective trials or have poor predictive ability.
TABLE 25-4 Prognostic Indicators for Severe Acute Pancreatitis
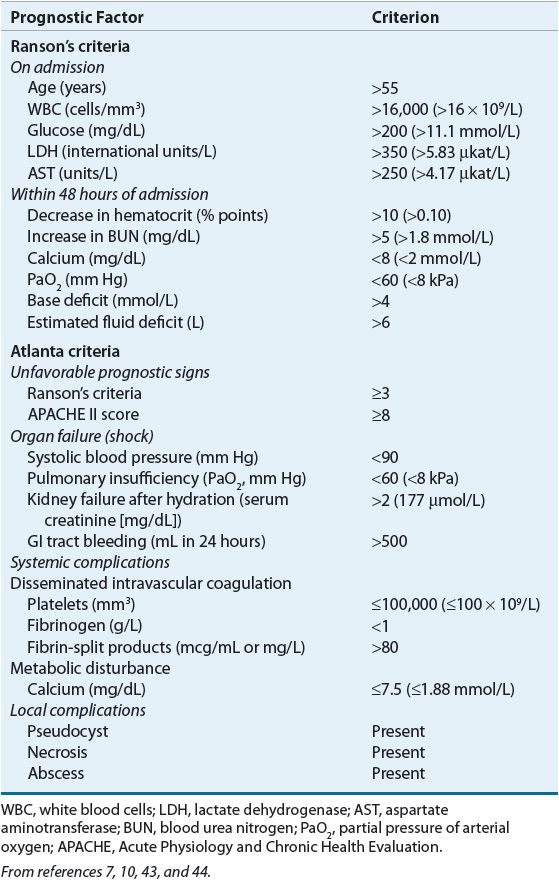
The first scoring system developed for pancreatitis was the Ranson’s criteria. It assesses 11 variables that must be monitored at the time of admission and during the initial 48 hours of hospitalization.10 Severe acute pancreatitis is characterized by three or more criteria. While still used by many clinicians, the Ranson’s score does not correlate well with disease severity. The Atlanta scoring system was developed based on consensus opinions; it consolidates clinical indicators, organ failure, and local complications to provide an ongoing assessment of disease severity.7
The Acute Physiology and Chronic Health Evaluation II (APACHE II) system uses 12 indicators of physiologic and biochemical function, age, and previous health status to predict mortality in critically ill patients, but it is not specific to pancreatitis. The APACHE II score is calculated within the first 24 hours and is considered among the best predictors of severity on admission. A score greater than or equal to 8 points is associated with an increased risk of organ failure and mortality.10 Other scoring systems include the Bedside Index of Severity in Acute Pancreatitis (BISAP), the Harmless Acute Pancreatitis Score (HAPS), and a computer-based tool using blood urea nitrogen (BUN), pleural effusion, and serum calcium. SIRS criteria alone are sensitive for predicting organ failure and death, but are not specific to severe acute pancreatitis.1,44
Clinical Course and Prognosis
The clinical course of acute pancreatitis varies from a mild transitory disorder to a severe necrotizing disease. Mild acute pancreatitis is self-limiting and subsides spontaneously within 3 to 5 days. Mortality is influenced by etiology, as idiopathic and postoperative acute pancreatitis have higher rates than gallstone- or alcohol-related disease. First and second occurrences also carry a higher mortality than subsequent episodes. Mortality increases with unfavorable early prognostic signs, local complications, and organ failure. Persistent organ failure is a greater risk than transient organ failure.3 Severe pancreatitis with either organ failure or infected necrosis is associated with a mortality of approximately 30%, which increases when both are present.3 Death during the first few days results from SIRS and multiorgan failure. When death occurs after this period, it is usually a result of infected necrosis, pancreatic abscess, and sepsis.10
TREATMENT
Acute Pancreatitis
Desired Outcome
Treatment of acute pancreatitis is aimed at relieving abdominal pain and nausea, replacing fluids, correcting electrolyte, glucose, and lipid abnormalities, minimizing systemic complications, and preventing pancreatic necrosis and infection. Management varies depending on the severity of the attack (Fig. 25-3). Patients with mild acute pancreatitis respond very well to the initiation of supportive care and the reduction of pancreatic secretions. Patients with severe acute pancreatitis should be treated aggressively and monitored closely.
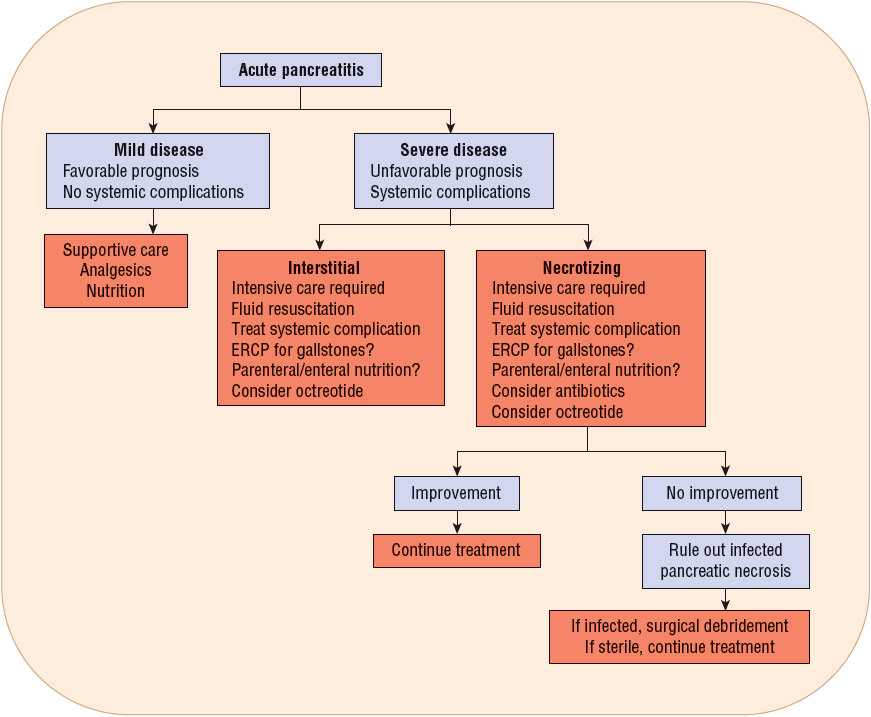
FIGURE 25-3 Algorithm of guidelines for evaluation and treatment of acute pancreatitis. (ERCP, endoscopic retrograde cholangiopancreatography.)
General Approach to Treatment
All patients with acute pancreatitis should receive supportive care, including IV fluid resuscitation, adequate nutrition, and effective relief of pain and nausea. The use of nasogastric aspiration offers no clear advantage in patients with mild acute pancreatitis, but it is beneficial in patients with profound pain, severe disease, paralytic ileus, and intractable vomiting.10 Patients predicted to follow a severe course will require treatment of cardiovascular, respiratory, renal, and metabolic complications.1 Aggressive fluid resuscitation is essential to correct intravascular volume depletion.45 Patients with pancreatitis and SIRS should be treated according to SIRS guidelines. IV potassium, calcium, and magnesium are used to correct electrolyte deficiency states. Insulin is used to treat hyperglycemia. Local complications resolve as the inflammatory process subsides. However, patients with necrotizing pancreatitis may require antibiotics and surgical intervention.10 Medications listed in Table 25-2 should be discontinued if possible ![]() .
.
Nonpharmacologic Therapy
Nonpharmacologic therapy includes ERCP for removal of any underlying biliary tract stones, surgery, and nutritional support. Surgery is indicated in patients with pancreatic pseudocyst or abscess or to drain the pancreatic bed if hemorrhagic or necrotic material is present. The need for admission to an intensive care unit should also be addressed. Advances in minimally invasive surgical techniques are changing practice with respect to timing and approach to managing infected necrotizing pancreatitis, and may help lower the risk of mortality in the most critical patients.2,11,46,47
Nutrition and Probiotics
Nutritional support plays an important role in the management of patients with mild or severe disease as acute pancreatitis creates a catabolic state that promotes nutritional depletion. This can impair recovery, increase the risk of complications, and prolong hospitalization.48,49 Patients with mild acute pancreatitis can begin oral feeding when bowel sounds have returned and pain has resolved.7 In severe or complicated disease, nutritional deficits develop rapidly and are complicated by tissue necrosis, organ failure, and surgery. Nutritional support should begin when it is anticipated that oral nutrition will be withheld for more than 1 week.50 In the past, there was concern that enteral feeding stimulated pancreatic enzyme secretion and exacerbated the underlying disease. However, a Cochrane Collaboration review that included eight randomized controlled trials found that enteral nutrition results in decreased morality, multiple organ failure, and need for surgical intervention compared with parenteral nutrition.50 Possible mechanisms for this include protection of the gut barrier and prevention of colonization with pathogenic bacteria, both of which may prevent translocation of bacteria and infection.11 Therefore, the enteral route is preferred over the parenteral in patients with severe acute pancreatitis provided that it can be tolerated. Ongoing trials are addressing some remaining issues such as the optimal timing to initiate enteral feeding and the safety of the nasogastric route as compared with nasojejunal. If enteral feeding is not possible or if the patient is unable to obtain sufficient nutrients, total parenteral nutrition should be implemented before protein and calorie depletion become advanced. IV lipids should not be withheld unless the serum triglyceride concentration is greater than 500 mg/dL (5.65 mmol/L).10
Clinical trials do not support the use of probiotics in the treatment of acute pancreatitis as they have not shown a benefit. One prospective randomized trial in patients with predicted severe acute pancreatitis showed an increase in mortality with probiotics compared with placebo.51
Pharmacologic Therapy
Recommendations
Patients with acute pancreatitis often require IV antiemetics for nausea. Those with severe acute pancreatitis should be treated with antisecretory agents to prevent stress-related mucosal bleeding. Patients also require appropriate fluid resuscitation and pain management, but there is controversy surrounding both of these therapies. Octreotide has been studied as a specific therapy in severe acute pancreatitis, but its efficacy remains uncertain (see Fig. 25-3). Prophylactic antibiotics used to be widely used, but clinical trials have failed to identify a group of patients that benefit from this therapy.
Fluid Resuscitation
Vasodilation from the inflammatory response, vomiting, and nasogastric suction contribute to hypovolemia and fluid and electrolyte abnormalities, thus necessitating replacement. Evidence for the benefit of adequate fluid resuscitation comes from observational studies demonstrating an associated increase in morbidity and mortality with failure to improve laboratory indicators of hemoconcentration (i.e., hematocrit and BUN). There are no large randomized trials to provide specific recommendations. Guidelines call for rapid replacement of fluid, without details on rate or type of fluid ![]() .
.
Observational studies have identified both benefit (decreased mortality and organ failure) and harm (abdominal compartment syndrome) associated with early aggressive fluid administration. Most studies have compared standard therapy with aggressive fluid therapy over the first 24 hours retrospectively. One trial found that administration during the first 24 hours of at least one third the cumulative volume given over the first 72 hours was associated with a decrease in mortality.52 A similar trial found a decrease in SIRS, organ failure at 72 hours, and length of stay in patients who received more fluid during the first 24-hour period than subsequent 24-hour periods.53 In contrast, another study found that patients who received more than 3.1 L of fluid during the first 24 hours had higher rates of persistent organ failure, respiratory failure, and renal failure than those who received smaller volumes.54 In a prospective, randomized trial, goal-directed fluid replacement therapy of 3 mL/kg/h for the first 20 hours did not result in a reduction in SIRS or C-reactive protein (CRP).55 Replacement at rates of 10 to 15 mL/kg/h was associated with more abdominal compartment syndrome, mechanical ventilation, and sepsis in the first 2 weeks following presentation than standard therapy in another trial.56
Interpretation of these trials is complicated by the likelihood that sicker patients were given larger volumes of fluid. Studies of fluid resuscitation in acute pancreatitis suggest that some patients may not require aggressive fluid resuscitation, while others may require gradual fluid administration. For example, those with reduced cardiac reserve may do better if fluid is replaced over 72 hours rather than 24 to 48 hours.57
In addition to questions about the rate and volume of fluid that should be administered to patients with acute pancreatitis, there is also debate regarding which fluid is most appropriate. A small randomized trial found that goal-directed resuscitation with lactated Ringer’s produced a reduction in SIRS and CRP at 24 hours compared with normal saline.55 The study protocol used aggressive replacement with a bolus of 20 mL/kg of lactated Ringer’s followed by 150 to 300 mL/h for the first 24 hours. If patients responded to this therapy as assessed by BUN, the rate could be reduced to 2 mL/kg/h. Patients with SIRS or sepsis should be resuscitated according to sepsis guidelines ![]() .58
.58



