Table 27-1. Prescribing Recommendations for Neuropathic Pain
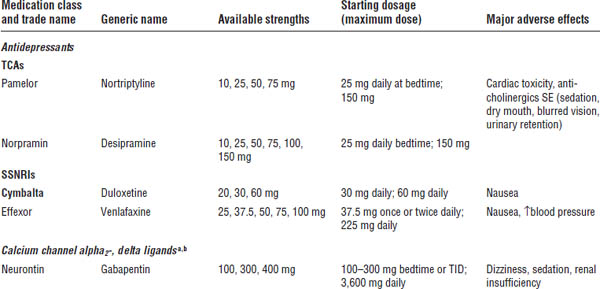

Adapted from Dworkin RH, O’Connor AB, Audette J, et al., 2010; O’Connor AB, Dworkin RH, 2009.
Boldface indicates one of top 100 drugs for 2012 by units sold at retail outlets, www.drugs.com/stats/top100/2012/units.
SSNRI, selective serotonin and norepinephrine reuptake inhibitor; TCA, tricyclic antidepressant.
a. First-line treatment.
b. Consider lower starting doses and slower titration in the elderly.
c. Second-line treatment, may be appropriate as first-line treatment in certain circumstances.
Chronic pain assessment should include a detailed history of the pain’s intensity and characteristics, a physical examination emphasizing the neurological exam, and a psychosocial assessment (Figure 27-1).
The purpose of diagnostic tests, such as x-rays, computed tomography, or magnetic resonance imaging scans, or laboratory tests differs depending on the type of pain. In cancer patients, the major purpose of diagnostic testing is to visualize the disease progression. In chronic benign pain, the major purpose of diagnostic testing is to rule out the presence of any diseases for which there is a curative treatment.
Figure 27-1. Algorithm for Comprehensive Evaluation and Management of Chronic Pain
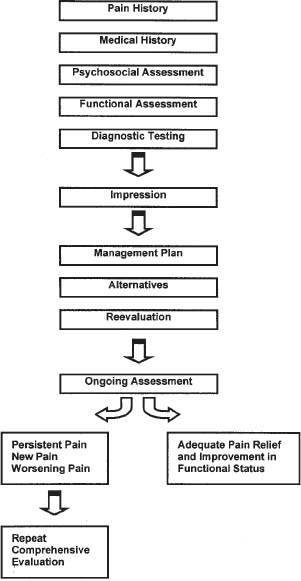
Reproduced with permission from the Pain Management Center of Paducah.
Goals of Pain Management
Acute pain
The goal in acute pain management is to provide patients with pain relief that allows them to rest comfortably and allows postsurgery or postinjury rehabilitation. This goal can be accomplished by administering short-acting medications as needed.
Malignant pain
A major goal of cancer pain management is to relieve the patient’s pain without inducing disabling side effects.
The World Health Organization (WHO) has developed a three-step hierarchy for analgesic pain management in cancer pain patients (Figure 27-2). In general, this program includes using nonopioid analgesics as a baseline, supplementing with opioid analgesics as needed, and adding adjunctive medications when appropriate.
Cancer patients may suffer from constant pain that continues for months or years. For this reason, treatment with long-acting agents is more appropriate than treatment with short-acting medications. However, short-acting agents, referred to as “breakthrough” or “rescue” doses, are often available in addition to the long-acting medications.
Chronic benign pain
The goal of chronic benign pain treatment is to restore the patient to the highest degree of function possible or a decrease in pain intensity of at least 30%.
Multimodal therapy, the use of several different types of treatment, is usually required. Multimodal therapies include nerve blocks, rehabilitation, physical therapy, pharmacotherapy, acupuncture, and psychotherapy. Basic pharmacotherapy follows the WHO guidelines for treating cancer pain (Figure 27-2).
Figure 27-2. The WHO’s Three-Step Hierarchy for Analgesic Pain Management in Cancer Patients
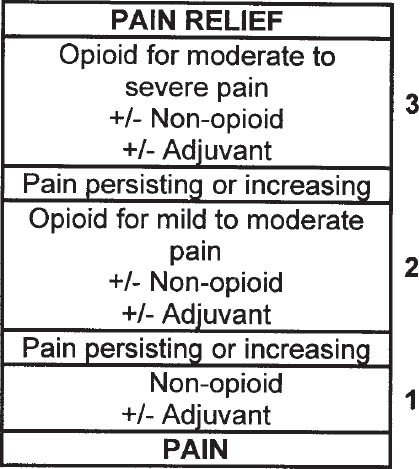
Reproduced with permission from WHO 1996.
Analgesics are categorized into nonopioid analgesics, opioid analgesics, and adjuvant analgesics. Nonopioid analgesics, such as acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs), relieve all types of mild to moderate pain. Prescription products containing acetaminophen are limited to 325 mg per dosage unit because of the potential for severe liver failure. NSAIDs are commonly used as part of the treatment regimen for pain associated with inflammation and cancer-related bone pain. Unless contraindicated, all pain patients should first be given a trial of nonopioid analgesics. Nonopioids and opioids relieve pain via different mechanisms. Thus, combination therapy offers the potential for improved relief with decreased doses and fewer side effects. Nonopioids do not produce tolerance, physical dependence, or addiction (see Table 27–2).
Adjuvant analgesics are drugs with a primary indication other than pain. Commonly used analgesic adjuvants include antiepileptic drugs, tricyclic antidepressants, local anesthetics, and SNRIs.
Principles of opioid use
Opioids have no ceiling effect of analgesia.
Oral medications should be used whenever possible. Intramuscular injections are painful and should be avoided.
When patients have constant or near-constant pain, analgesics should be given around the clock. Long-acting opioid analgesics are often used for this purpose.
The use of short-acting opioids as rescue medication is controversial in chronic benign pain. If allowed, doses of rescue medications should range from 10% to 15% of the total daily long-acting opioid dose.
Table 27-2. Common Nonopioid Analgesics
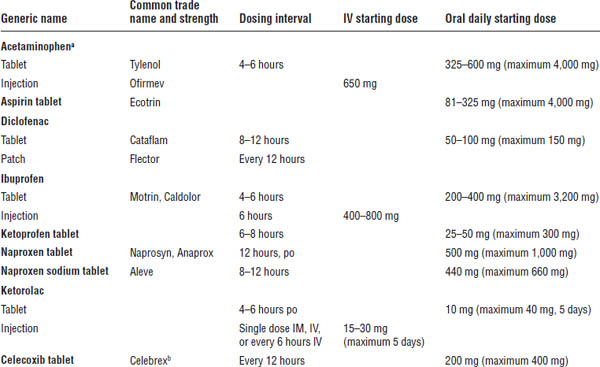
Adapted from Baumann TJ, Strickland JM, Herdon CM, et al., 2011.
a. Rare but serious skin reactions have been reported.
b. One of top 100 drugs for 2012 by units sold at retail outlets, www.drugs.com/stats/top100/2012/units.
Mixed agonist–antagonist opioids are not used in chronic pain. They may induce a withdrawal syndrome in patients tolerant to opioids.
Drug Therapy
Mechanism of action
Morphine and other opioid agonists are thought to produce analgesia by mimicking the action of endogenous opioid peptides that bind at opioid receptors in the antinociceptive pathway.
Opioid receptors are located in the central nervous system (CNS), pituitary gland, and gastrointestinal (GI) tract. They are abundant in the periaqueductal gray matter of the brain and the dorsal horn of the spinal cord, two areas that are very active in pain reduction.
When a drug binds to one of these receptors as an agonist, it produces analgesia. When a drug binds to one of these receptors as an antagonist, analgesia and other effects are blocked.
The three major types of opioid receptor sites involved in analgesia are mu (μ), delta (δ), and kappa (κ):
■ Binding to the μ receptor produces analgesia, sedation, euphoria, respiratory depression, physical dependence, constipation, and other effects.
■ Activation of δ receptors produces analgesia without many adverse events. However, there is no available δ-receptor agonist.
■ Activation of the κ receptor produces analgesia and respiratory depression. In addition, psychotomimetic effects such as anxiety, strange thoughts, nightmares, and hallucinations are common.
Opioid analgesics
Opioids are classified by activity at the receptor site; that is, they are classified as pure opioid agonists, agonist–antagonists, or pure opioid antagonists.
Pure opioid agonists primarily activate μ receptors, although they may produce some κ-receptor activation (Table 27-3). Pure opioid agonists are the most clinically useful opioid analgesics.
Morphine is the prototypical pure opioid agonist. Methadone is an opioid agonist with additional antagonist activity at the NMDA (N-methyl-D-aspartate) receptor. The NMDA receptor is believed to be active primarily in chronic pain.
Mixed agonist–antagonists bind as agonists at the κ receptor, producing weak analgesia. They bind as weak antagonists at the μ receptor (Table 27-4). The result is more dysphoria and psychotomimetic effects with a lower risk of respiratory depression.
Pentazocine is the prototypical agonist–antagonist opioid. Buprenorphine is actually a partial agonist at μ and κ receptors. This opioid has limited efficacy in pain management and is primarily used in detoxification programs.
Opioid antagonists block the μ and κ receptors (Table 27-5). These drugs do not produce analgesia. They are used to reverse respiratory and CNS depression caused by overdose with opioid agonists. Naloxone and naltrexone are opioid antagonists.
Opioid analgesic adverse effects
Central nervous system
Opioids produce a number of CNS effects, including sedation, euphoria, dysphoria, changes in mood, and mental clouding. Confusion, disorientation, and cognitive impairment are also possible.
Chronic sedation can be treated with CNS stimulants such as methylphenidate or dextroamphetamine. Modafinil also promotes daytime wakefulness.
Mild to moderate muscle jerks, known as myoclonus, are common in patients on high doses of opioids. Myoclonus can be treated by changing the opioid dose, changing the opioid, or giving low doses of a benzodiazepine.
Neuroendocrine
Morphine acts in the hypothalamus to inhibit the release of gonadotropin-releasing hormone and corticotropin-releasing factor, thus decreasing levels of luteinizing hormone, follicle-stimulating hormone, adrenocorticotropic hormone, and β-endorphins.
Changes in hormone levels may cause decreased levels of testosterone and cortisol, disturbances in menstruation, and sexual dysfunction.
High doses of morphine and related opioids produce convulsions. Most convulsions occur at doses far in excess of those required to produce analgesia.
Respiratory
Respiratory depression is the most serious opioid-induced adverse effect. Opioids depress respiration by a direct effect on the brain-stem respiratory centers, making the brain stem less responsive to carbon dioxide.
The μ receptor is the primary receptor involved in respiratory depression, although activation of the κ receptor also contributes.
Table 27-3. Starting Doses for Strong Opioids for Severe and Moderate to Severe Pain in Adults: Mu Agonists
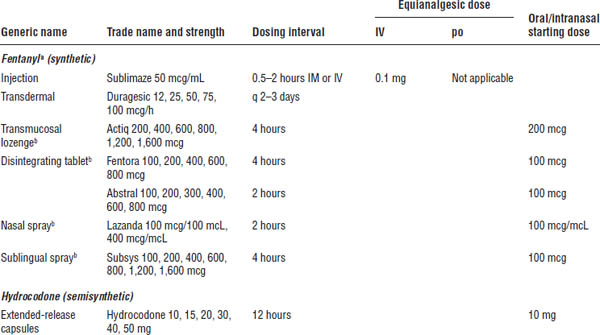


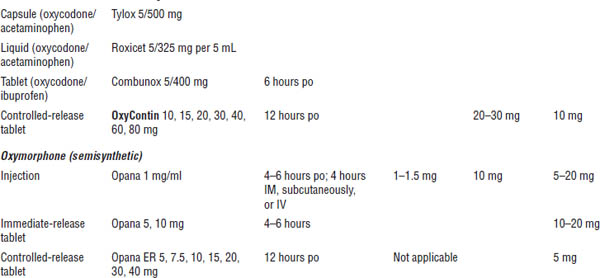
a. One of top 100 drugs for 2012 by units sold at retail outlets, www.drugs.com/stats/top100/2012/units.
b. For use in breakthrough cancer pain.
Table 27-4. Opioid Dosing for Mild to Severe Pain in Adults
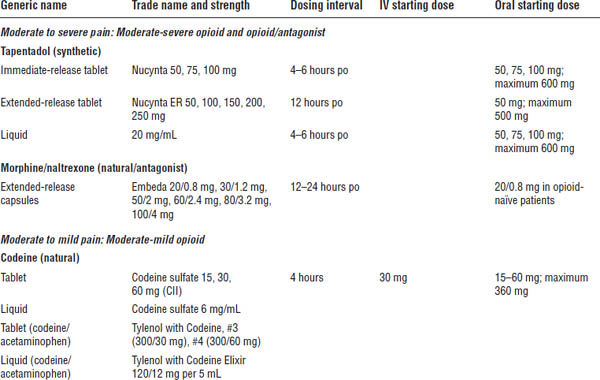


a. One of top 100 drugs for 2012 by units sold at retail outlets, www.drugs.com/stats/top100/2012/units.
At equianalgesic doses, all pure opioid agonists depress respiration to the same degree. The agonist–antagonists have a ceiling effect (i.e., a dose beyond which no further respiratory depression or analgesia is produced), but this level is usually above recommended doses.
Opioids depress cough by inducing a direct effect on the cough reflex in the medulla.
Cardiovascular
Therapeutic doses of many opioids produce peripheral vasodilation, reduced peripheral resistance, and inhibition of the baroreceptor reflexes.
Peripheral vasodilation results primarily from opioid-induced release of histamine. Orthostatic hypotension and fainting can result. The naturally occurring and semisynthetic products are potent histamine releasers. Fentanyl has little propensity to release histamine.
Table 27-5. Opioid Antagonists
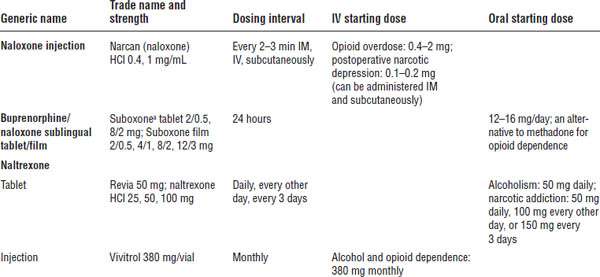
a. One of top 100 drugs for 2012 by units sold at retail outlets, www.drugs.com/stats/top100/2012/units.
Methadone has been associated with torsades de pointes, an atypical rapid ventricular tachycardia, at an average daily dose of 400 mg. Methadone should be used cautiously in patients on other QTc-prolonging medications. Patients should receive an electrocardiogram prior to therapy, at day 30, and once yearly while receiving methadone.
Gastrointestinal
All clinically significant μ agonists produce some degree of nausea and vomiting by direct stimulation of the chemoreceptor trigger zone in the medulla, sensitization of the vestibular system, and slowing of GI motility.
Nausea and vomiting commonly occur in ambulatory patients (28% and 15%, respectively). Both can be pretreated with an antiemetic such as promethazine or prochlorperazine.
Opioids promote constipation by delaying gastric emptying, slowing bowel motility, and decreasing peristalsis. Opioids may also reduce secretions from the colonic mucosa. At its worst, GI dysfunction results in ileus, fecal impaction, and obstruction.
Because transdermal delivery bypasses absorption from the GI tract, constipation has been reported to be less frequent with this delivery method than with other methods.
Patients on opiates do not develop tolerance to constipation. All patients taking around-the-clock opioid analgesics should be placed on prophylactic bowel regimens. Bowel regimens include increased fluid and fiber intake, daily stool softeners, and mild laxatives.
Severe constipation is managed with osmotic laxatives such as magnesium citrate and milk of magnesia.
Genitourinary
Opioids increase smooth muscle tone in the bladder and ureters and may cause bladder spasm and urgency.
An opioid-induced increase in urethral sphincter tone can make urination difficult. Urinary retention is most common in elderly men.
Biliary
Opioids increase smooth muscle tone in the biliary tract, especially in the sphincter of Oddi, which regulates the flow of bile and pancreatic fluids. This effect can result in a decrease in biliary and pancreatic secretions and a rise in the bile duct pressure. Patients may experience epigastric distress and occasionally biliary spasm.
All opioids are capable of causing constriction of the sphincter of Oddi and the biliary tract. Use caution when prescribing an opioid to a patient with biliary tract disease and pancreatitis.
Skin and eye
Therapeutic doses of morphine dilate cutaneous blood vessels, which causes flushing on the face, neck, and upper thorax. Sweating and pruritus may also occur. These changes may be caused in part by the release of histamine. Histamine release may induce or worsen asthmatic attacks in predisposed patients and can lead to wheezing, bronchoconstriction, and status asthmaticus.
Skin rash around the transdermal fentanyl patch is a common side effect caused by the patch adhesive.
Following a toxic dose of μ agonists, miosis is evident but insufficient alone to confirm a definitive diagnosis of opioid intoxication.
Overdose
Acute overdose with opioids is manifested by respiratory depression; somnolence progressing to stupor or coma; skeletal muscle flaccidity; cold, clammy skin; constricted pupils; and sometimes pulmonary edema, bradycardia, hypotension, and death.
An opioid antagonist such as naloxone may be given to block opioid receptors and reverse the effects of overdose.
Antagonist administration may cause a complete reversal of opioid effects and precipitate an acute withdrawal syndrome in persons physically dependent on opioids.
Antagonists are dosed to patient response every few minutes. If no response is observed after administration of 15 mg of naloxone, the diagnosis of opioid-induced respiratory depression should be questioned.
Tolerance and physical dependence
The use of opioids is often limited by concerns regarding tolerance, physical dependence, and addiction.
Tolerance can be defined as a state in which a larger dose is required to produce the same response that could formerly be elicited by a smaller dose. Tolerance to analgesia is demonstrated by the need for an increased dosage of a drug to produce the same level of analgesia. Tolerance is sometimes mistaken for disease progression in cancer patients.
Tolerance to adverse effects of opioids occurs after weeks of continuous administration. Tolerance to the constipating and neuroendocrine effects of opioids does not occur.
Physical dependence is the occurrence of a withdrawal syndrome after an opioid is stopped or quickly decreased without titration. Warn patients to avoid abrupt discontinuation of such drugs.
Addiction is a behavior pattern involving the continued use of a substance for nonmedical reasons despite harm. It is characterized by impaired control over drug use, compulsive use, craving, and continued use despite harm.
Pharmacokinetics of Selected Opioids
Morphine
Compared with other opioids, morphine is relatively insoluble in lipids (e.g., in adults, only small amounts of the drug cross the blood–brain barrier).
Morphine does not accumulate in tissues when given in normal doses and therefore does not cause increasing toxicity with frequent dosing.
Morphine is primarily metabolized by glucuronidation during the first pass through the liver. Approximately 50% of morphine is converted by the liver to morphine-3-glucuronide and 15% to morphine-6-glucuronide (M6G). The pharmacologic effects of morphine (both analgesia and side effects) are in part caused by M6G.
Much of an oral dose is inactivated during this first pass through the liver; consequently, oral doses need to be much larger than parenteral doses to produce the same analgesic effects.
Fentanyl
Fentanyl is highly soluble in lipids. It accumulates in skeletal muscle and fat and is released slowly into the blood. Plasma half-life is 3–4 hours after parenteral administration.
Fentanyl is rapidly metabolized, primarily by dealkylation, to inactive metabolites in the liver. This process is mediated through the cytochrome P450 (CYP450) 3A4 hepatic enzyme system. The presence of inactive metabolites makes fentanyl a preferred drug in patients with liver dysfunction.
Fentanyl is not used orally because of low oral bioavailability.
Transdermal fentanyl
The uptake of fentanyl through the skin is relatively slow and constant. The skin does not metabolize the drug, and 92% of the dose is delivered into the bloodstream as intact fentanyl.
Because of temperature-dependent increases in fentanyl release from the patch system as well as increased skin permeability, an increase in body temperature to 40°C (104°F) theoretically may increase serum fentanyl concentrations by approximately one-third.
Fentanyl is absorbed into the upper layers of the skin, forming a depot. Fentanyl then becomes available to systemic circulation. Serum fentanyl concentrations are measurable within 2 hours after application of the first patch, and analgesic effects can be observed 8–16 hours after application. Steady state is reached after several sequential patch applications.
Transmucosal fentanyl citrate lozenge
The absorption pharmacokinetics of fentanyl from the oral transmucosal dosage form is a combination of initial rapid absorption from buccal mucosa and delayed absorption of fentanyl from the GI tract. Normally 25% of the total dose is available by buccal absorption, and 25% is available from the GI tract, making the total bioavailability 50%.
Analgesia begins in 10–15 minutes, peaks in 20 minutes, and persists for 1–2 hours.
Transmucosal fentanyl is indicated only for those who are already receiving and tolerant to around-the-clock opioid therapy.
Fentanyl citrate buccal tablet
Following buccal administration, fentanyl is readily absorbed with an absolute bioavailability of 65%. Approximately 50% of the total dose administered is absorbed transmucosally and becomes systemically available. The remaining half of the total dose is swallowed and undergoes more prolonged absorption from the GI tract.
Buccal tablets are indicated only for those already receiving around-the-clock opioid therapy.
Fentanyl buccal soluble film
The pharmacokinetic absorption involves a combination of rapid absorption from the buccal mucosa and delayed absorption from the GI tract. Absorption from the buccal mucosa is 51% of the total dose, and the remaining 49% of the dose is absorbed from the GI tract. Approximately 20% of the dose absorbed from the GI tract is available for systemic absorption.
Fentanyl soluble film is indicated for breakthrough pain in cancer patients 18 years of age and older who are opioid tolerant and receiving around-the-clock opioid therapies.
Fentanyl sublingual spray
The fentanyl sublingual spray displays a mean absolute bioavailability of 76%. Its bioavailability is dependent on the amount of the dose that is absorbed through the sublingual mucosa and the amount swallowed from the gastrointestinal tract. Variable increases in fentanyl plasma concentration have occurred in patients with mucositis.
Fentanyl nasal spray
Intranasal fentanyl is absorbed from the nasal mucosa and its bioavailability is approximately 20% higher than that of transmucosal fentanyl citrate. The plasma concentration is dose dependent and increases in a linear manner across the dose range. A maximum plasma concentration is reached 15–21 minutes after a single dose.
Methadone
After therapeutic doses, about 90% of methadone is bound to plasma protein and is widely distributed in tissues. Methadone is found in low concentrations in the blood and the brain, with higher concentrations in the kidney, spleen, liver, and lung. Terminal half-life is extremely variable (15–55 hours); therefore, accumulation is possible, and dosing intervals need to be carefully monitored.
Methadone is extensively metabolized in the liver, mainly by N-demethylation. This process appears to be mediated primarily by CYP450 3A4 and to a lesser extent by CYP450 2D6. The major metabolites are excreted in the bile and urine.
Analgesic efficacy does not correspond to the half-life of the drug. Methadone may be dosed every 3 hours for pain control.
Oxycodone
Oxycodone is metabolized to noroxycodone, oxymorphone, and their glucuronides via the CYP450 enzyme system. The major circulating metabolite is noroxycodone. Noroxycodone is reported to be a weaker analgesic than oxycodone. Oxymorphone, although possessing good analgesic activity, is present in the plasma only in low concentrations. Its metabolism is mediated by CYP450 2D6.
Hydromorphone
Hydromorphone is metabolized to three major metabolites: hydromorphone 3-glucuronide, hydromorphone 3-glucoside, and dihydroisomorphine 6-glucoside. Whether hydromorphone is metabolized by the CYP450 system is not known. Hydromorphone is a poor inhibitor of CYP450 isoenzymes and is not expected to inhibit the metabolism of other drugs.
Meperidine
Normeperidine, a toxic metabolite of meperidine, produces anxiety, tremors, myoclonus, and generalized seizures when it accumulates with repetitive dosing. Patients with compromised renal function and concomitant use of benzodiazepines are particularly at risk. Naloxone does not reverse this hyperexcitability. For these reasons, meperidine should not be used for more than 48 hours in patients with renal or CNS disease or at doses greater than 600 mg every 24 hours.
Tapentadol
Tapentadol is metabolized to its major metabolite, tapentadol-O-glucuronide via glucuronidation and two minor metabolites, N-desmethyl tapentadol and hydroxy tapentadol, via the CYP450 enzyme system. The metabolism via CYP450 is not as significant as the phase 2 conjugation. Approximately 97% of the parent drug is metabolized. The major metabolic pathway is via conjugation with extensive metabolism through phase 2 pathways and minor metabolism by phase 1 oxidative pathways. The metabolites do not produce any analgesic activity. The bioavailability is approximately 32% after a single dose because of first-pass metabolism. Peak serum concentrations are typically observed 1.25 hours after a dose.
Drug Interactions and Drug–Disease Interactions
Drug interactions
All drugs with CNS-depressant actions (barbiturates, benzodiazepines, alcohol) can intensify sedation and respiratory depression caused by morphine and other opioids.
Antihistamines, tricyclic antidepressants, and atropine-like drugs can exacerbate morphine-induced constipation and urinary retention.
Antihypertensive drugs and others that lower blood pressure can exacerbate opioid-induced hypotension.
The combination of Demerol, Exalgo, Abstral, or Nucynta with a monoamine oxidase inhibitor (MAOI) may produce a syndrome characterized by excitation, delirium, hyperpyrexia, convulsions, and severe respiratory depression. Death may also occur. Although this syndrome has not been reported with other opioids, combinations containing opioids and MAOIs should be avoided. Patients should not take an opioid medication within 14 days of taking an MAOI.
Agonist–antagonists can precipitate a withdrawal syndrome if administered to an individual who is physically dependent on a pure opioid agonist.
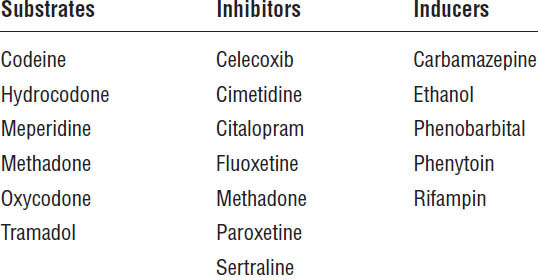
CYP450 enzymes metabolize codeine, hydrocodone, fentanyl, methadone, and oxycodone. Although not well documented, drug interactions through this system may exist. In particular, fentanyl and oxycodone should be used with caution in a patient on a CYP450 3A4 inhibitor. The patient should be monitored over an extended period and dose adjustments made as appropriate.
Codeine, hydrocodone, and oxycodone require metabolism through CYP450 2D6 to an active drug (Table 27-6). Approximately 7% of Caucasians, 3% of African Americans, and 1% of Asians are poor metabolizers of CYP450 2D6; they produce no CYP450 2D6 or produce undetectable levels of it. Poor metabolizers may experience little or no analgesia from drugs requiring CYP450 2D6 for conversion to active metabolites.
About 5% of the patients have multiple copies of the CYP450 2D6 gene, making them ultrafast metabolizers. The clearance of some opioids may be increased, making more frequent dosing of the medications necessary.
Drug–disease interactions
In view of the extensive hepatic metabolism of opioids, their effects may be increased in patients with liver disease, particularly those with severe liver failure. Most opioids require dose reduction in severe liver disease.
Fentanyl, morphine, and methadone require dosing adjustment in renal impairment. Doses of fentanyl and morphine should be reduced 25% when creatinine clearance (CrCl) is 10–50 mL/min and by 50% when CrCl is < 10 mL/min. The dosing interval of methadone should be increased to at least every 6 hours when CrCl is 10–50 mL/min and to every 8 hours when CrCl is < 10 mL/min.
Table 27-6. CYP450 2D6 Enzyme Activity
Renal impairment slows the clearance of morphine conjugates, resulting in accumulation of the active metabolite M6G. For this reason, dosage reduction may be advisable in the presence of clinically significant renal impairment.
Methadone appears to be firmly bound to protein in various tissues, including the brain. After repeated administrations, methadone gradually accumulates in tissues. The risk of accumulation is greater in patients with impaired renal or hepatic function because both organs are involved in the metabolism of methadone.
Patient Counseling
Respiratory depression is increased by concurrent use of other drugs with CNS-depressant activity (e.g., alcohol, barbiturates, benzodiazepines). Outpatients should be warned against the use of alcohol with all other CNS depressants.
Constipation is commonly experienced with short- and long-term use of opioids. Suggest that patients take a stool softener and mild laxative if constipation occurs during the course of treatment. Inform patients about symptoms of hypotension (e.g., lightheadedness, dizziness). Patients should minimize hypotension by moving slowly when changing from a supine to an upright position.
Advise patients that opioids are drugs of potential abuse and should never be taken by anyone other than the person for whom they are prescribed. Patients should never adjust the dose of their medication without first consulting their health care provider.
Liquid formulations should be measured with an appropriate dose-measuring cup or spoon and not a regular teaspoon or tablespoon.
Combinations should not exceed the maximum daily dose of acetaminophen (4 g/day, no liver damage present) to prevent liver toxicity.
Women who are pregnant or planning to become pregnant should consult their health care provider before starting therapy.
Fentanyl transdermal patch
The fentanyl transdermal patch must be applied to a clean, nonhairy site on the upper torso. Only water should be used to clean the area. Soap or alcohol can increase the effects of the medication and should not be used. The patch should not be applied to oily, broken, burned, cut, or irritated skin. It must be held in place for a minimum of 30 seconds to ensure adhesion.
Each new patch should be applied to a different area of skin to avoid irritation. If a patch comes off or causes irritation, it should be removed and a new patch applied to a different site.
To dispose of the patch, fold it in half and flush down the toilet.
Do not cut or damage the patch.
Temperature-dependent increases in fentanyl release from the patch could result in an overdose. Advise patients to avoid exposing the patch to direct external heat sources such as heating pads, electric blankets, heat lamps, saunas, hot tubs, and heated waterbeds. In addition, patients who develop a high fever while wearing the patch should contact their health care provider immediately.
Long-acting opioid formulations
The long-acting formulations should be swallowed whole (i.e., not broken, chewed, or crushed).
Avinza, a long-acting morphine capsule formulation, contains fumaric acid. Doses above 1,600 mg per day contain a quantity of fumaric acid that has not been demonstrated to be safe and may result in serious renal toxicity.
Embeda (extended-release morphine sulfate and naltrexone hydrochloride) contains pellets of an extended-release oral formulation of morphine sulfate surrounding an inner core of naltrexone hydrochloride. Embeda is usually prescribed for pain when drug abuse or diversion is a concern. The capsules should be swallowed whole, or the capsule contents may be sprinkled on applesauce. Crushing, dissolving, or chewing the pellets could block the analgesic effects of morphine by causing an increased release of naltrexone and also could cause a rapid release of a potentially fatal morphine dose. Alcohol consumption may also increase the release and absorption of morphine, leading to an overdose of morphine.
Kadian and Avinza (long-acting morphine sulfate) may be opened and the beads ingested with a small amount of applesauce (sprinkle administration). In addition, Kadian is approved for sprinkle administration through a gastrostomy tube.
Patients must not consume alcoholic beverages or any medications containing alcohol while on Opana ER therapy. The co-ingestion of alcohol with Opana ER may result in increased plasma levels and a potentially fatal overdose of oxymorphone. In addition, food increases the Opana ER maximum concentration by approximately 50%. Opana ER should be ingested 1 hour before and 2 hours after a meal.
Transmucosal fentanyl citrate lozenge
The lozenge is used by placing it in the mouth between the cheek and the gum. Consumption of the lozenge should take 15 minutes. Another lozenge may be used 30 minutes after the start of the first one. Tell the patient not to bite or chew the lozenge.
To dispose of a finished lozenge, discard the handle in a place that is out of reach of children and pets. If medicine remains on the handle, place the handle under hot running tap water until the medicine is dissolved. Never leave unused or partly used lozenges where children or pets can get to them.
Fentanyl citrate buccal tablet
Once removed from the blister pack, the lozenge must be used right away. It is placed in the mouth above the back molars and between the upper cheek and gum. It is left in place until it dissolves, which may take between 14 and 25 minutes. After 30 minutes, any remaining tablet is swallowed with a glass of water.
Parameters to monitor
Evaluate for pain control 1 hour after opioid administration. If analgesia is insufficient, consider a dosage increase. Patients taking opioids chronically should be evaluated regularly for adequate doses.
Monitor the patient for respiratory depression. Higher risk for respiratory depression exists in patients who are not tolerant to opioid analgesics. Consider treatment when the respiratory rate is less than 8–12 respirations per minute for 30 minutes or longer despite stimulation or if oxygen saturation is less than 90%.
If a patient is easily arousable, he or she is unlikely to have respiratory depression.
Tramadol
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


