Other Nonneoplastic Disorders
Paul W. Biddinger
AMIODARONE-INDUCED THYROID DISEASE
Amiodarone is an iodinated drug used for the treatment of cardiac arrhythmias. Although an effective drug for arrhythmias, amiodarone can produce side effects in various organs including the thyroid. The effects range from subclinical abnormalities of thyroid function tests to overt thyroid hypofunction or hyperfunction.1,2 Most patients receiving amiodarone remain euthyroid. The reported incidence of amiodarone-induced hypothyroidism (AIH) usually ranges from 10% to 20%, whereas the incidence of amiodarone-induced thyrotoxicosis (AIT) ranges from 5% to 10%.2
Pharmacology
Amiodarone is a benzofuran derivative with two atoms of iodine per molecule, and its structure bears resemblance to thyroid hormones (Fig. 6.1). Iodine constitutes 37.5% of the molecular weight of amiodarone. Metabolism releases free iodine from about 10% of the bodily content of amiodarone per day. Typical maintenance doses in the 200 to 400 mg range thus yield about 7 to 15 mg of iodine per day, far in excess of the World Health Organization’s recommended daily adult intake of 0.15 mg.3
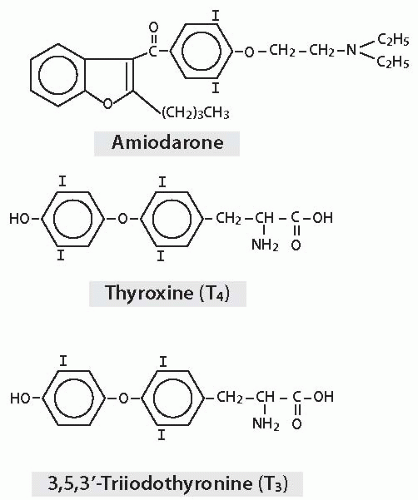 FIGURE 6.1. Structures of amiodarone, thyroxine (T4), and triiodothyronine (T3). |
Amiodarone is amphiphilic and distributes widely in the body. Its high lipid solubility results in storage in various sites including adipose tissue, liver, lung, myocardium, skeletal muscle, kidney, thyroid, and brain. Amiodarone is slowly released with a half-life in the range of 1 to 3 months and thus can have effects long after therapy is discontinued.1,4,5
Effect on Thyroid Follicular Cells and Hormone Synthesis
The release of excessive iodine results in reduction of iodine oxidation by the thyroid and decreased thyroid hormone synthesis, an adaptive phenomenon known as the Wolff-Chaikoff effect.6 Generally, the thyroid escapes from this inhibition within a couple of weeks.7 Amiodarone and its main metabolite, desethylamiodarone, appear to have a direct toxic effect on follicular cells, although the release of excessive iodine may be a significant factor.8 A potential indirect mechanism of amiodarone injury is the promotion of thyroid autoimmunity. However, most studies have shown that amiodarone is unlikely to be associated with an increase in thyroid autoantibodies if the individual lacked them before therapy.1
Effect on Thyroid Hormone Metabolism
Amiodarone decreases the peripheral conversion of thyroxine (T4) to 3,5,3′-triiodothyronine (T3) and 3,3′,5′-triiodothyronine (rT3) to 3,3′-diiodothyronine (T2) because of its inhibition of 5′-deiodinase (5′-D).9,10 Amiodarone also inhibits uptake of T4, T3, and rT3 by peripheral tissues.11 This results in the elevation of serum T4 and rT3 and the decrease of serum T3 in patients receiving amiodarone, although these levels may still remain within the normal range. Serum thyroid-stimulating hormone (TSH) may be elevated during the early months of therapy, but it generally returns to normal.12
Amiodarone-Induced Hypothyroidism
AIH is more prevalent in areas with high dietary iodine intake.1,2,13 Studies tend to show a slight female predominance with a ratio of 1.5:1.1,2 About two-thirds of patients have preexisting thyroid disease, whereas the remaining one-third start with apparently normal glands (Table 6.1).1 The preexisting thyroid disease is usually autoimmune (Hashimoto) thyroiditis. Patients with preexisting thyroperoxidase (TPO) and/or thyroglobulin antibodies are at higher risk to develop AIH when treated with amiodarone.1,14,15 AIH may be transient or persistent. Although 10% to 20% of patients develop AIH in the short term, the prevalence appears to decrease to 5% to 10% after a year of therapy.2 Persistence is usually associated with an underlying disease such as autoimmune thyroiditis.16
Several pathogenic mechanisms appear to be involved in AIH. One is failure to escape from the Wolff-Chaikoff effect. This effect is usually transient, but those with AIH may have a defect in hormonogenesis
that prevents escape.15 The defect in hormonogenesis may be related to preexisting thyroid disease, particularly autoimmune thyroiditis. Direct and indirect cytotoxic mechanisms may also be involved. However, at this time, the pathogenic mechanisms remain unclear, particularly for those individuals lacking preexisting thyroid disease.
that prevents escape.15 The defect in hormonogenesis may be related to preexisting thyroid disease, particularly autoimmune thyroiditis. Direct and indirect cytotoxic mechanisms may also be involved. However, at this time, the pathogenic mechanisms remain unclear, particularly for those individuals lacking preexisting thyroid disease.
Table 6.1 Comparison of AIH and AIT | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pathology
The pathologic findings of AIH are not well documented in the medical literature. Preexisting thyroid disease is probably the predominant finding in most cases. Extrapolation from animal studies suggests that most cases would show various ultrastructural changes of the follicular cells such as increased lysosomes, lysosomal inclusion bodies, evidence of necrosis and apoptosis, lipofuscin production, and markedly dilated endoplasmic reticulum.17
Amiodarone-Induced Thyrotoxicosis
AIT is more prevalent in areas with low dietary iodine intake.1 AIT tends to have a higher frequency among men.2,16 Two forms of thyrotoxicosis are associated with amiodarone therapy (Table 6.1). Type 1 is caused by excessive production and release of thyroid hormone and is usually associated with an underlying thyroid disease such as multinodular goiter or Graves disease (diffuse toxic hyperplasia).1,2 Type 1 AIT is a manifestation of the Jod-Basedow phenomenon characterized by hyperthyroidism because of an increased supply of iodine.2,18 Type 2 AIT is caused by a destructive thyroiditis that develops in thyroid glands without preexisting disease.1,2 Type 2 AIT currently appears to be the more common of the forms.2,19 Ultrasonic assessment typically shows elevated color Doppler flow in type 1 AIT, whereas it shows decreased color Doppler flow in type 2 AIT.20 The differentiation of the two types of AIT can be challenging because there are cases with mixed or indefinite features.2,21 Differentiating whether a case is “pure” type 1 AIT or a mixture of both types can be difficult.
Pathology
The pathologic changes associated with type 1 AIT are predominantly those of the underlying thyroid disease. Type 2 AIT is characterized by involution features in most of the thyroid parenchyma with scattered foci of follicular destruction.22,23 Grossly, the cut surfaces have a homogenous dark red to reddishbrown gelatinous appearance, reflective of abundant colloid (Fig. 6.2). The disrupted follicles contain detached follicular cells, foamy macrophages, and lymphocytes (Fig. 6.3). Sometimes all that remains of a damaged follicle is a solid collection of macrophages. Multinucleated giant cells also may be present in areas of follicular disruption. Fibrosis may be associated with the areas of follicular damage. The intact follicles are lined predominantly by flat epithelial cells and contain abundant thick colloid, consistent with involution because of low plasma TSH. Some follicular cells exhibit cytoplasmic vacuolization.
Treatment and Prognosis
Treatment of AIH is supplementation with oral thyroxine if amiodarone is necessary for the control of the cardiac arrhythmia. If amiodarone therapy can be discontinued, most patients will return to a euthyroid state, particularly if they did not have a preexisting thyroid disorder. Those patients with autoimmune thyroiditis are less likely to achieve normal thyroid function.
Treatment of AIT can be challenging. Therapy for type 1 AIT is directed at blocking the synthesis of thyroid hormones and decreasing the uptake of iodine by the thyroid.1 Methimazole or
another thioamide is generally used to diminish hormone synthesis, whereas potassium perchlorate is used to decrease iodine uptake. In contrast, glucocorticosteroid therapy is used for type 2 AIT. The anti-inflammatory effects of steroids are often effective in ameliorating the destructive thyroiditis characteristic of type 2 AIT.
another thioamide is generally used to diminish hormone synthesis, whereas potassium perchlorate is used to decrease iodine uptake. In contrast, glucocorticosteroid therapy is used for type 2 AIT. The anti-inflammatory effects of steroids are often effective in ameliorating the destructive thyroiditis characteristic of type 2 AIT.
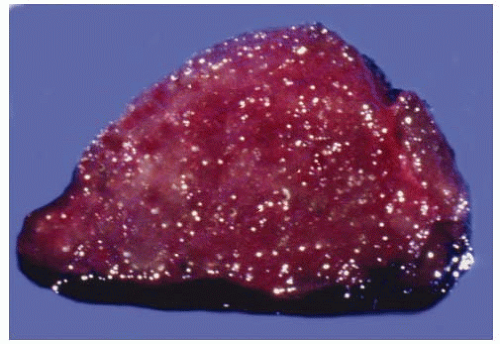 FIGURE 6.2. Cross-section of thyroid with AIT, type 2. The gland has a gelatinous appearance because of abundant colloid. |
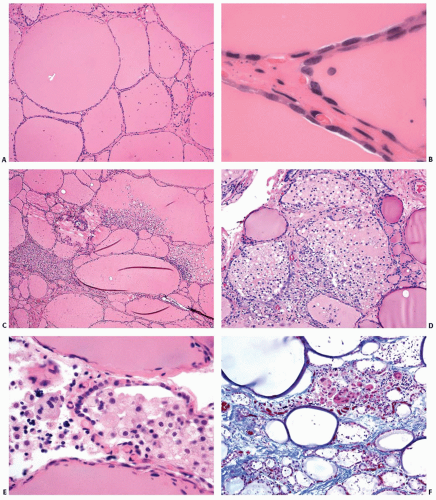 FIGURE 6.3. Representative photomicrographs of thyroid with AIT, type 2. Most thyroid follicles are intact and show involution features (A and B). Focal follicular disruption with infiltration and replacement of colloid by foamy macrophages is shown in images (C-F). A high-power view of focal follicular disruption is shown in image (E) with multinucleated giant cells admixed with the macrophages. Focal fibrosis in the region of damaged follicles is shown in image (F) (trichrome stain). |
Definitive treatment of type 1 AIT is directed at the underlying thyroid disorder, which may require radioablation or thyroidectomy. Cessation of amiodarone therapy will eventually allow most type 2 AIT patients to return to a euthyroid state, but this may not be a satisfactory option for individuals with life-threatening arrhythmias refractory to other medications. Thyroidectomy is a consideration if amiodarone must be continued and medical control of thyrotoxicosis is unsuccessful.
MINOCYCLINE-ASSOCIATED CHANGES (“BLACK THYROID”)
Dark brown to black pigment can accumulate in the thyroid after long-term use of minocycline, a long-acting antibiotic of the tetracycline group. The degree of pigmentation can be sufficient to give the thyroid a diffusely black gross appearance.
Pathogenesis
The composition of the pigment and mechanism of formation are not entirely clear, but numerous studies indicate that the pigment derives from oxidation of minocycline by TPO.24,25,26 The pigment appears to be a unique polymeric product with many properties similar to melanin and lipofuscin. The pigment accumulates within the lysosomes of the follicular cells.27,28
In vitro studies have shown minocycline to be an inhibitor of TPO-catalyzed iodination.26,29 The mechanism is consistent with substrate inhibition of TPO-catalyzed iodination of tyrosyl residues in thyroglobulin.29 The reaction may also generate a reactive intermediate metabolite, and thus, there is the possibility of associated damage to follicular cells and impaired thyroid function. However, the potential for clinically significant toxicity appears to be very low given the scarcity of cases with abnormal thyroid function. A study in which human volunteers were given high doses of minocycline for up to 85 days failed to reveal abnormal pigmentation in thyroid biopsies.24 This suggests that one or more other factors may be involved in the pathogenesis of pigment formation.
Clinical Presentation
Most cases are discovered coincidentally when a thyroid lesion is excised surgically because of a nodular lesion or the thyroid is examined at the time of autopsy. The incidence of minocycline-associated thyroid pigmentation is unknown, but it appears to be quite low given the small number of reported cases and the large number of individuals who have received the drug since its introduction in 1967. However, cases may go unrecognized because of normal thyroid function and absence of gross distortion, and individual cases may no longer gain publication in contemporary medical literature.
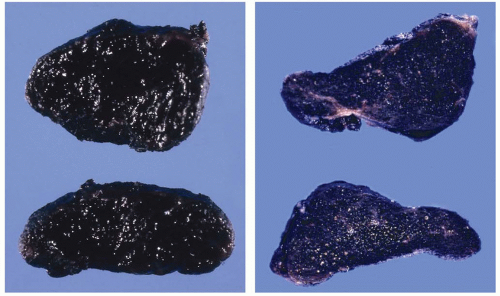 FIGURE 6.4. Two separate cases of black pigmentation of thyroid associated with minocycline therapy.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|