cortical bone (9). In ischemic necrosis, initially fat necrosis and death of hematopoietic cells are noted, followed in several weeks by loss of osteocytes. Characteristically, the bone looks acellular, with empty ghostlike osteocytic lacunae and no evidence of osteoblast surface activity (Fig. 14.2). Osteonecrotic tissue often shows evidence of past remodeling such as creeping substitution or juxtaposition of new bone, suggesting that following the initial ischemia the bone-forming potential is stimulated. Most probably, the length of the ischemic episode dictates the extent to which, if possible, remodeling and new formation can occur. Obviously, terminal ischemia, that is, complete disruption of the only arterial supply, results in irreversible death. It has been estimated that, with ischemia, in less than 2 hours the changes that occur in osteocytes are probably reversible, but most likely irreversible damage occurs after 4 hours (10). Correlation with light microscopic features has been made (Table 14.3).
TABLE 14.1 Classification of Osteonecrosis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
space, and aggregation of interchromatin granules and the development of large interruptions on the plasma membrane and actual extrusion of cytoplasm. These changes are not detectable by light microscopy. However, Catto (8,9) has studied osteonecrosis following femoral neck fractures. Within 24 hours of injury, fibrin and hemorrhage are the most obvious changes. With ensuing days, there is a proliferation of capillaries, with foamy macrophages appearing about day 4. New bone formation is not usually evident at the fracture site in less than 5 days. However, active osteoblast formation and juxtaposed new bone formation are noticeable by the second week. The hematopoietic tissue shows loss of nuclear detail within days after death has occurred and complete loss of nuclei with cloudy-like appearance 2 weeks afterward. In humans, osteocytic nuclei begin to disappear after 4 to 5 days, but are more obvious 2 weeks later. In a rabbit model of bone ischemia, James and Steijn-Myagkaya (11) showed irreversible damage at 24 hours and empty lacunae after 24 hours. Despite the slow loss of osteocytic nuclei, traditionally a hallmark of osteonecrosis, the bone is most likely dead after the vascular injury and long before all cellular detail is lost. Therefore, the pathologist interpreting histopathologic sections, relying solely on cellular criteria of osteocytic empty lacuna spaces, may underdiagnose the presence of osteonecrosis.
TABLE 14.2 Clinicopathologic Sites and Syndromes Associated with Osteonecrosis | |
|---|---|
|
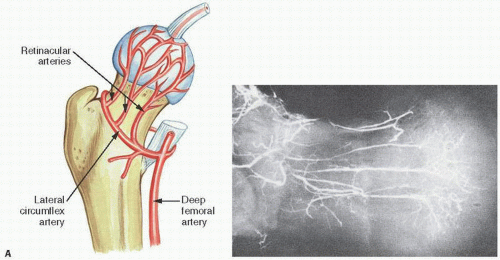 FIGURE 14.1. Vascularity of the femoral head. Specimen radiograph (A) and gross femoral head (Continued) |
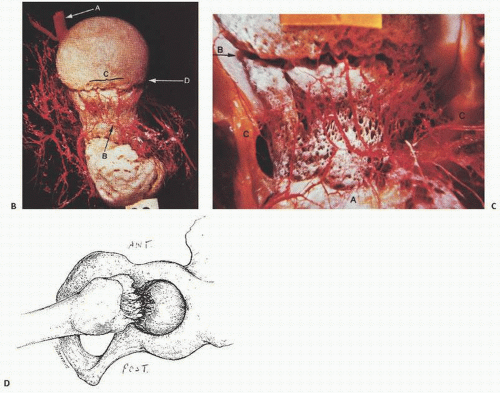 FIGURE 14.1. (Continued) (B) (A, medial femoral artery; B, cervical arteries; C, subcapital zone; D, site of subcapital fracture) and (C) (A, cervical branches; B, site of subcapital fractures; C, lateral circulation) with arterial injection showing fine tenuous arterial circulation to the femoral head, a site at great risk for disruption at the time of subcapital fractures (D). (After Crock HV. A revision of the anatomy of the arteries supplying the upper end of the human femur. J Anat. 1965;99:77-88.) |
TABLE 14.3 Timetable of Microscopic Bone Changes in Osteonecrosis | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
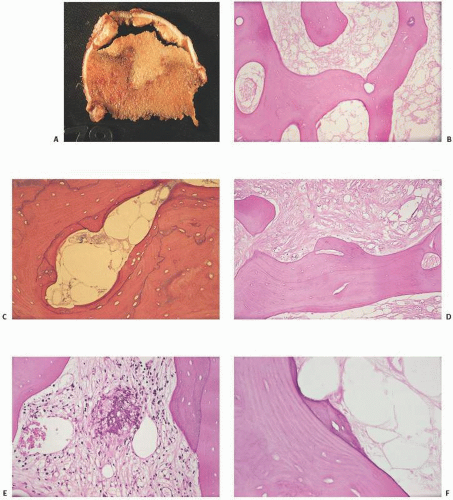 FIGURE 14.2. Dead bone. Dead bone has a gross (A) and microscopic pallor (B) (lack of staining intensity). The bone characteristically lacks nuclei in osteocyte lacunar spaces (B, C) and, until phagocytic activity ensues, also lacks surface remodeling by osteoblasts and osteoclasts. The marrow simultaneously undergoes necrosis with loss of nuclei of fat or marrow cells, and a foggy appearance ensues followed by fibrosis of marrow (D) or even saponification (calcification of the fat) (E). Attempts at repair include new bone apposition. This creeping substitution may be jagged (F) or smooth (Continued) |
 FIGURE 14.2. (Continued) (G). Eventually, the bone is resorbed with large osteoclast-type resorption spaces (H). Thrombi may be observed in osteonecrosis, giving credibility to thrombophilia as a significant etiologic event (I). In some cases, ischemic marrow and microfractures support a link between subchondral insufficiency fractures and osteonecrosis (J). Infarcted bone and fat may be associated with a bluish discoloration of the mineralized tissue (K, L). |
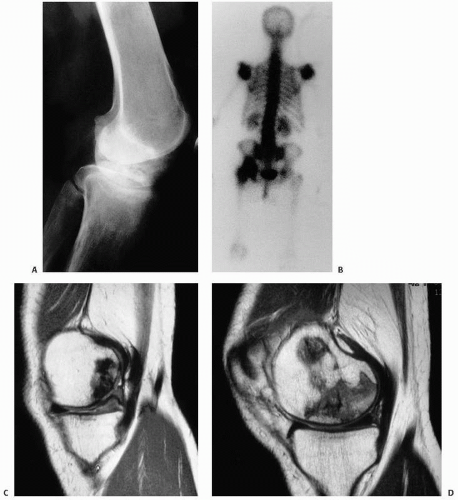 FIGURE 14.3. Roentgenographic, bone scan, and magnetic resonance imaging (MRI) findings in osteonecrosis. Fuzzy, irregular radiopacities are typical of late-stage osteonecrosis on routine x-rays, distal femoral condyle (A). Bone scan is hot, due to both hypervascularity and new bone production in hip (B). (C-F) Areas of necrosis in the medial femoral condyle as demonstrated on MRI images; all images are fast-spin echo. C and D are proton density (TR 4,000; TE 30/Ef). (Continued) |
prediction of the likelihood of collapse (13). The classic pattern of osteonecrosis of the femoral head is a circumscribed ovoid or crescent-shaped low signal in subchondral bone, the rim representing the reparative zone between normal and ischemic damaged bone. A second inner rim, the “double lesion sign,” may be seen as a high signal on T2 and is thought to be pathognomonic of osteonecrosis by some (13).
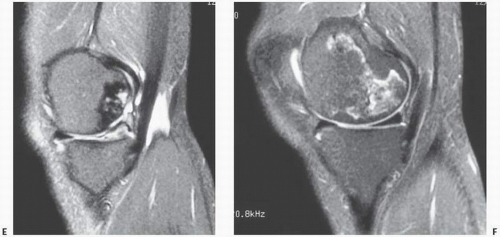 FIGURE 14.3. (Continued) E and F are fat-suppressed T2-weighted (TR 3,500; TE 39 Ef). Images C and E demonstrate damage of the articular cartilage superficial to the area of necrosis. |
TABLE 14.4 Causes of Osteonecrosis of the Femoral Head in One Large Series | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
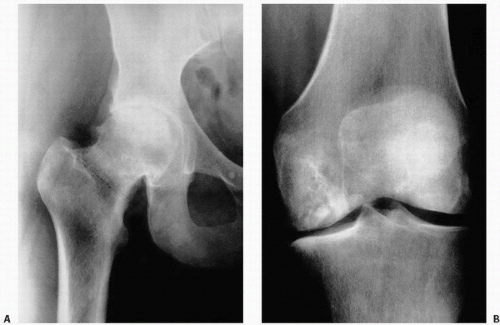 FIGURE 14.4. Avascular necrosis of the femoral head (A) and knee (B). Irregular radiodense subarticular portions of the femoral head and knee indicate dead (and partially reparative) bone. |
TABLE 14.5 Stages of Osteonecrosis Based on Roentgenographic Studies | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
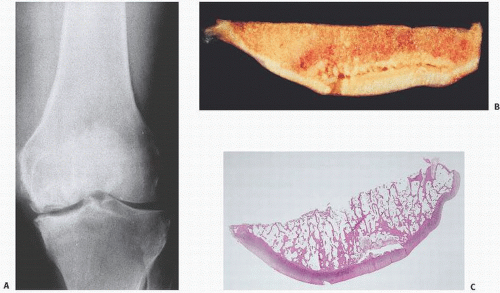 FIGURE 14.5. Avascular necrosis of the knee. Anteroposterior x-ray of the knee in a middle-aged woman with sudden onset of knee pain. Sclerosis and disruption of the medial femoral condyle is seen (A). A coronal section [(B) gross; (C) microscopic] taken through the medial condyle of a patient with osteonecrosis of the knee shows a zone of bone necrosis immediately under the articular surface characterized by an opaque-yellow appearance. Immediately beyond the necrotic zone is a band of hyperemia. (Continued) |
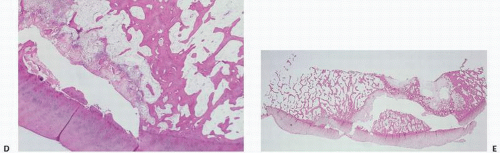 FIGURE 14.5. (Continued) Separating the necrosis bone from the overlying cartilage is a gap created by the collapse of the bone trabeculae in the necrotic segment. Microscopically, there is collapse and compression of the dead subchondral bone. A radiolucent space ensues (C, D). Dead bone may adhere to residual viable cartilage and eventually dislodge into the joint, manifesting itself as a loose body (E). |
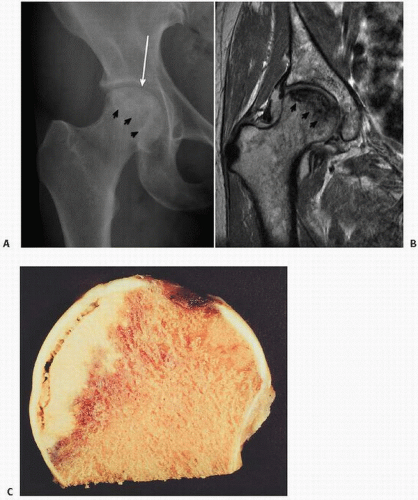 FIGURE 14.6. Avascular necrosis of the femoral head. Roentgenogram (A) and coronal MRI image (B) demonstrate avascular necrosis of the femoral head seen as density sclerosis in A and low signal in B (black arrows). Note the crescent sign (long arrow in A indicating subchondral collapse, corresponding to the lack of perfect sphericity of the femoral head in B. (Continued) |
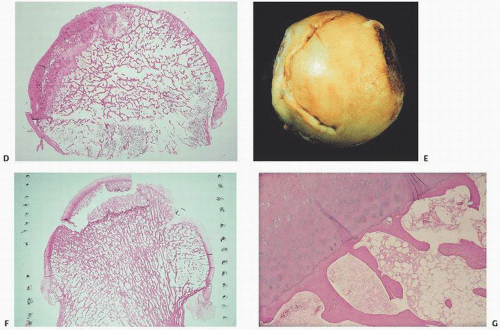 FIGURE 14.6. (Continued) Coronal section showing crescenteric zone of opaque yellow dead bone with radiolucent subchondral zone [(C) gross; (D) microscopic]. The zone of dead bone may or may not be evident on gross examination, depending on whether the cartilage has collapsed and broken off. The femoral head cartilage is dimpled (E) and fractured (F). Microscopically, there is a dislodged fragment of viable cartilage and adherent dead bone (G). |
Magnetic resonance imaging is the most sensitive and specific diagnostic method.
Symptomatology, although important for planning treatment strategies for individual patients, is not a reliable indicator of disease severity because it is quite variable; thus, it should not be part of the classification system.
A major determinant of prognosis is whether the femoral head is at the precollapse or postcollapse stage.
Size of lesion is important for prognosis; however, the best method of assessing the size of the lesion has yet to be fully elucidated or defined.
Acetabular articular cartilage involvement is an important determinant of disease severity in classification systems. Again, the method to determine this, whether preoperatively or with intraoperative inspection, has not been fully elucidated.
The amount of femoral head depression is a major prognostic indicator of disease progression.
Measurement of the amount of the depressed femoral head that is involved as well as the length of the crescent may not be reproducible.
The location of the lesion may not be as important as size when dealing with large lesions, because lateral lesions are usually large. Medial lesions, although quite rare, are typically small and are associated with the best prognosis.
While small, medially located lesions have a low rate of progression, the natural history of asymptomatic medium-sized, and especially large, osteonecrotic lesions is progressive in most patients.
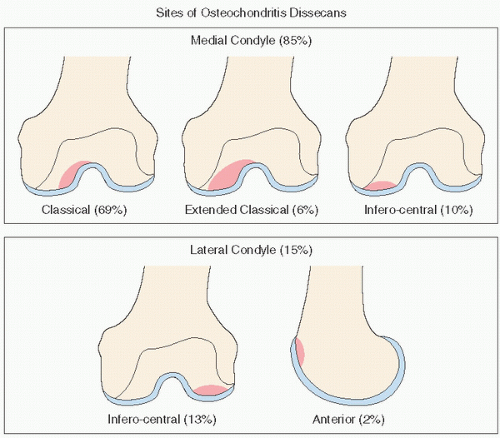 FIGURE 14.7. Osteochondritis dissecans: sites. |
population. Although destruction of the growth plate may ensue restricting blood flow to the epiphysis, and although the anatomical configuration of the femoral head may remain normal initially, eventual subchondral collapse may ensue with a flattened mushroom-shaped femoral head. Suggested factors determining the prognosis for any given patient include the patient’s age of onset, extent of involvement of the femoral head, loss of containment of the femoral head by the acetabulum in the weight-bearing position (subluxation), and a loss of motion of the hip joint. In the absence of subchondral fracture, AVN of the femoral head in children may resolve with no deformity. Legg-Calvé-Perthes disease is thought by some to be a disorder characterized by AVN, which, if complicated by pathologic subchondral fracture, may lead to further damage including resorption of bone and collapse of the femoral head.
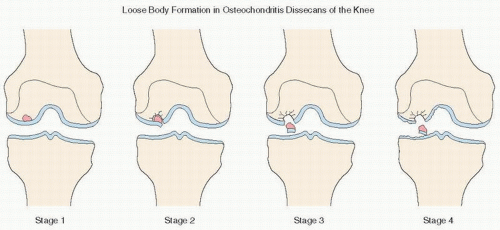 FIGURE 14.8. Osteochondritis dissecans: staging. |
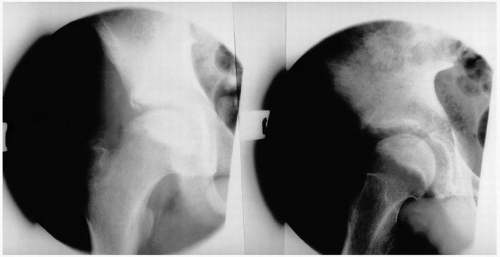 FIGURE 14.9. Legg-Calvé-Perthes disease. Roentgenographic features. |
osteonecrosis from fat embolization derives from biophysical forces. Fat has a greater viscosity than plasma, and its surface tension may facilitate adherence to arterial walls. It has been speculated that increased blood viscosity or increased arterial wall length or decreased vascular diameter as well as decreased intravascular pressure gradients all may reduce blood flow and potentiate intraosseous fat trigger damage. There are at least three proposed etiologies for fat embolization: the hyperlipidemia associated with fatty liver, endogenous production of lipoprotein, and the destruction of marrow fat. The latter may be a contributing factor in posttraumatic osteonecrosis.
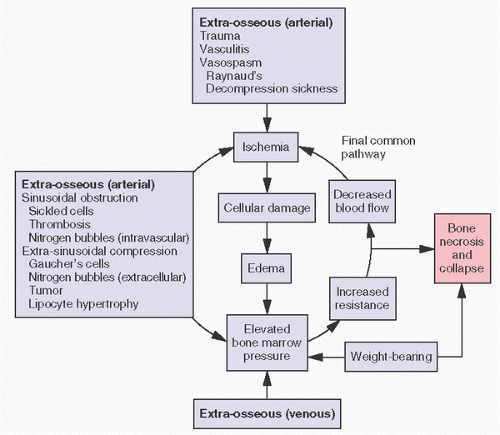 FIGURE 14.10. Proposed pathogenesis of osteonecrosis. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


