Fig. 19.1 The origin of pain and suffering.
Nociceptive pain is a defensive response to a variety of stimuli (e.g. mechanical, thermal or chemical) that activate nociceptor sensory units on nerve endings. It is defensive as it induces behaviour that avoids exacerbation of the pain, and allows us to protect a damaged part of the body while it heals. Painful stimuli are transmitted to the central nervous system (CNS) by two types of fibre. Fast fibres in the neospinothalamic pathways (mechanical and thermal stimuli) carry pain that is appreciated as sharp and localised, while slow fibres in the paleospinothalamic pathways (chemical stimuli) produce poorly localised aching, throbbing or burning pain (Fig. 19.2).
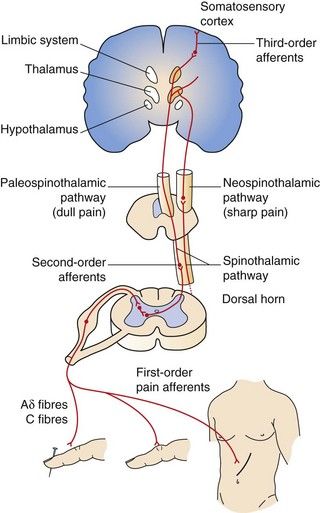
Fig. 19.2 Ascending pathways of pain perception.
Ascending pathways are activated following stimulation of afferent sensory nociceptive nerve terminals. Many mediators and neurotransmitters are involved during afferent stimulation of the nociceptive pathway. Mediator release (bradykinin, serotonin, prostaglandins) and thermal and mechanical influences can stimulate and sensitise the sensory nerve terminals of pain fibres, resulting in increased cation influx, depolarisation and generation of action potentials (see Fig. 19.3). Onward afferent transmission of ascending nerve impulses at the synapses in the dorsal horn involves transmitters such as substance P, glutamate and calcitonin gene-related peptide (CGRP) (Fig. 19.5). Hyperexcitability of pain fibres can also be promoted by other mediators. Prolonged activation of the nociceptive pathways can produce pathophysiological and phenotypic changes resulting in neuropathic pain that persists when the original pathological cause of the pain has resolved, and generation of nociceptive signals can occur at low levels of axonal stimulation.
Neuropathic pain may result from abnormal neuronal activity that persists beyond the time expected for healing of the injury. Examples include phantom limb pain following amputation and shingles causing pain well beyond the healing of the injury. There are multiple pathophysiological mechanisms underlying neuropathic pain. Neuropathic pain can be spontaneous (stimulus-independent), when it is usually described by the sufferer as shooting or lancinating sensations, electric shock-like pain or an abnormal unpleasant sensation (dysaesthesia). Alternatively, it can be an exaggerated response to a painful stimulus (hyperalgesia) or a painful response to a trivial stimulus (allodynia).
Some pain states have mixed nociceptive and neuropathic elements, for example mechanical spinal pain with local nerve damage such as radiculopathy or myelopathy.
The pathophysiological and molecular explanations for nociceptive and neuropathic pain and the endogenous responses that modulate pain are complex and incompletely understood. In brief, the genesis of nociceptive pain results from initial stimulation of afferent sensory neurons (nociceptors) by thermal, mechanical or chemical stimuli sufficient to stimulate the nociceptors (free nerve endings responsive to high-threshold noxious stimuli) on Aδ and C axons (Fig. 19.3). Nociceptors carry many surface receptors that modulate their sensitivity to stimulation, such as those for γ-aminobutyric acid (GABA), opioids, bradykinin, histamine, serotonin (5-hydroxytryptamine, 5-HT) and capsaicin, and mechanosensitive ion channels (responsive to touch, pressure, etc.). The particular type of painful stimulus may determine which receptors or ion channels are activated. Activation of the ion channels or receptors results in an influx of Na+/Ca2+ sufficient to generate action potentials in the nerve (Fig. 19.3). Most nociceptors are dormant (silent), but when tissue damage occurs nociceptors that were not normally active can be recruited and sensitise the area to painful stimuli. The afferent fibres from these nociceptors enter both the dorsal and ventral horns of the spinal cord, and link to ascending pathways. Chemical transmission of the afferent impulse travels across synapses in the spinal cord and in the projections of the ascending pathways to the thalamus, and from there to the reticular formation and the cortex. Transmitters in these central pathways include excitatory substances such as glutamate, tachykinins (neurokinins and substance P) and calcitonin gene-related peptide (CGRP). Synaptic transmission responsible for nociceptive pain can be reduced by local inhibitory neurons, mediated by GABA. There are also important descending modulatory pathways arising from the locus coeruleus that inhibit ascending pain pathways, with a further host of neurotransmitters including endogenous opioids, noradrenaline (acting on α2-adrenoceptors) and serotonin. Activation of these systems produces hyperpolarisation of neurons in the pain pathways (by inhibiting Na+/Ca2+ influx and enhancing K+ efflux) and prevents generation of nociceptive action potentials (Figs 19.4 and 19.5). The endogenous cannabinoids (anandamide and 2-arachidonyl glycerol) may also have modulatory functions in the descending pathways.
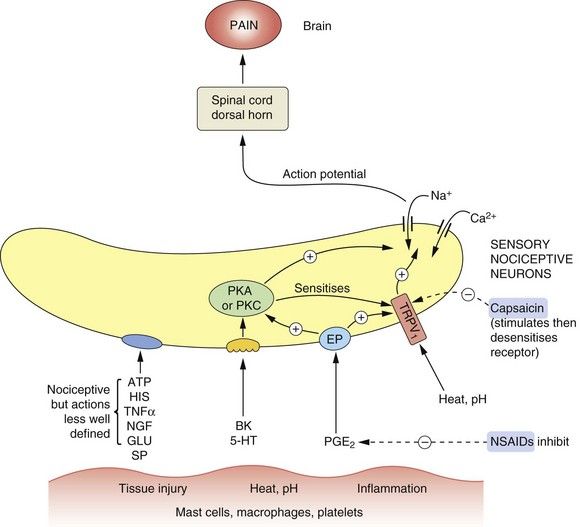
Fig. 19.3 Mediators involved in the genesis and modulation of pain.
Numerous mediators are able to stimulate or sensitise primary sensory neurons (nociceptors), leading to activation of nociceptive fibres. Tissue injury and other noxious stimuli such as heat or extremes of pH can stimulate the release of substances that act to promote pain, such as bradykinin (BK) and serotonin (5-hydroxytryptamine, 5-HT). Prostaglandin E2 (PGE2) acting at prostaglandin E receptors (EP) sensitises the nerve endings to the actions of nociceptive mediators including BK and 5-HT. ATP and histamine (HIS) also have nociceptive actions, acting in poorly defined ways. Heat and H+ stimulate the transient receptor potential vanilloid 1 (TRPV1) receptor, producing pain. The TRPV1 receptor is also sensitised by many other mediators. Capsaicin and other TRPV1 receptor stimulants desensitise the receptor on persistent stimulation, resulting in an analgesic effect. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the production of PGE2. Overall the effect of nociceptive stimuli is to depolarise the neuron, setting up action potentials in the fibres to the dorsal horn and pain-perceiving areas of the brain. Substance P (SP) and calcitonin gene-related peptide (CGRP) may also be involved in nociception. GLU, glutamate; NGF, nerve growth factor; PKA, protein kinase A; PKC, protein kinase C; TNFα, tumour necrosis factor α.
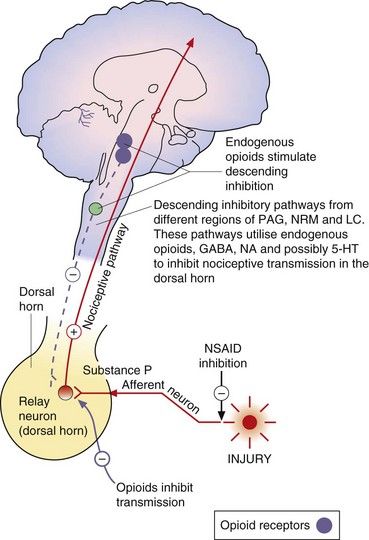
Fig. 19.4 Transmitters and receptors for pain perception and control.
The afferent nociceptive pathways are subject to inhibitory control. Opioids act at opioid receptor-rich sites in the periaqueductal grey matter (PAG), the nucleus raphe magnus (NRM) and other midbrain sites to stimulate descending inhibitory fibres that inhibit nociceptive transmission in the dorsal horn. Descending pathways from the locus coeruleus (LC) that are noradrenergic are also involved. Opioids also act at a local level in the dorsal horn (Fig. 19.5). Inhibitory modulation of nociceptive transmission via local nerve networks also results from actions of other agents (Fig. 19.5). 5-HT, 5-hydroxytryptamine (serotonin); GABA, γ-aminobutyric acid; NA, noradrenaline; NSAID, non-steroidal anti-inflammatory drug.
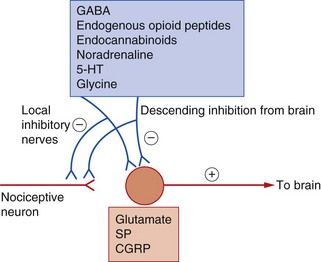
Fig. 19.5 Neurotransmitter substances involved in the genesis and modulation of pain.
Inhibitory modulation of nociception can occur via activation of the descending regulatory pathways and also by actions of local interneurons. 5-HT, 5-hydroxytryptamine (serotonin); CGRP, calcitonin gene-related peptide; GABA, γ-aminobutyric acid; SP, substance P.
Activation of pain pathways generates intracellular protein synthesis through stimulation of the proto-oncogene c-Fos and various inducible transcription factors in CNS neurons. These mediate the many changes in neuronal structure and the neuronal proliferation that occur with chronic pain.
Neuropathic pain may result from chronic hypersensitisation and phenotypic change in neurons. These changes can occur both peripherally and centrally, probably as a result of repetitive stimulation of synaptic transmission. In neuropathic and other chronic pain states, the following mechanisms may be important.
 Sensitisation of afferent inputs: this may include recruitment of silent nociceptors and a lower threshold for generation of action potentials in the spinal cord (through the action of cytokines released from glial cells), leading to enhanced action potential generation (Figs 19.3 and 19.5). Central sensitisation is also important with excessive neurotransmitter release. Abnormal voltage-gated Na+ channel expression may also be important in both peripheral and central sensitisation.
Sensitisation of afferent inputs: this may include recruitment of silent nociceptors and a lower threshold for generation of action potentials in the spinal cord (through the action of cytokines released from glial cells), leading to enhanced action potential generation (Figs 19.3 and 19.5). Central sensitisation is also important with excessive neurotransmitter release. Abnormal voltage-gated Na+ channel expression may also be important in both peripheral and central sensitisation. Dysfunctional descending pain-modulatory and -facilitatory pathways: the functioning of the descending modulatory pathway (Fig. 19.4) may become inadequate as a result of persistent nociceptive pain. Persistent painful stimuli may be related to the overactivity of endogenous pain facilitatory pathways that run in tracts from the medulla to the spinal dorsal horn. The imbalance in the pain pathways then maintains neuropathic pain states.
Dysfunctional descending pain-modulatory and -facilitatory pathways: the functioning of the descending modulatory pathway (Fig. 19.4) may become inadequate as a result of persistent nociceptive pain. Persistent painful stimuli may be related to the overactivity of endogenous pain facilitatory pathways that run in tracts from the medulla to the spinal dorsal horn. The imbalance in the pain pathways then maintains neuropathic pain states.
Analgesic drugs
Non-steroidal anti-inflammatory drugs (NSAIDs; Ch. 29) and opioids are the major classes of pain-relieving (analgesic) drugs. They act at different levels in the pain-transmitting pathways to influence the production and recognition of pain as indicated in Figures 19.3 and 19.4.
Non-steroidal anti-inflammatory drugs
These act mainly by blocking the peripheral generation of the nociceptive impulses. Prostaglandins enhance nociceptive impulses in peripheral afferent neurons by enhancing the ability of thermal, mechanical or chemical stimuli to increase Na+/Ca2+ influx and thereby to generate action potentials in nociceptive afferent neurons. NSAIDs inhibit the production of prostaglandins by the cyclo-oxygenase type 1 and type 2 (COX-1, COX-2) isoenzymes and thereby reduce the sensitivity of sensory nociceptive nerve endings to agents released by injured tissue that initiate pain, such as bradykinin and substance P. NSAIDs may also act on pain pathways in the CNS. Paracetamol, although not usually considered to be an NSAID, may also work in part through the same pathways. These drugs are considered fully in Chapter 29.
Opioids
These act on the spinal cord and limbic system, and stimulate the long descending inhibitory pathways from the midbrain to the dorsal horn. They produce their effects via specific receptors that are closely associated with the neuronal pathways which transmit pain from the periphery to the CNS.
Non-opioid, non-NSAID analgesics
A variety of drugs that were developed for other purposes are being used increasingly for their analgesic actions when NSAIDs and opioids are less effective, for example in neuropathic pain (see Table 19.2, below).
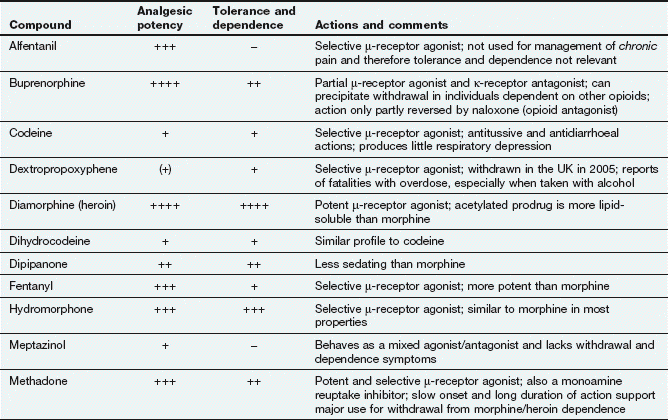
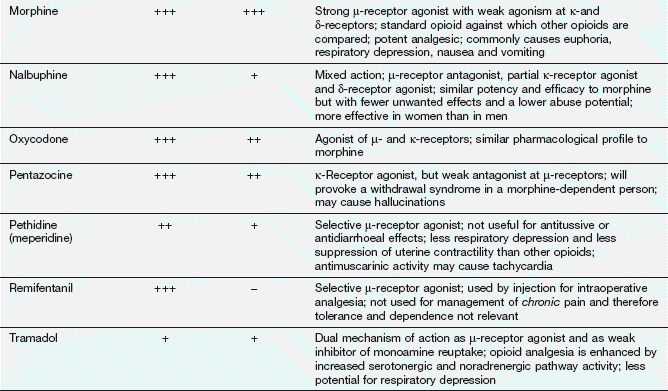
Opioid analgesics
Examples
buprenorphine, codeine, diamorphine (heroin), dihydrocodeine, fentanyl, methadone, morphine, oxycodone, tramadol
Opioid is a term used for both naturally occurring and synthetic molecules that produce their effects by an agonist action at opioid receptors. The terms opiate analgesic (specifically, a drug derived from the juice of the opium poppy, Papaver somniferum) and narcotic analgesic (which literally means a ‘stupor-inducing pain killer’) are no longer used.
Mechanism of action
The brain produces several endogenous opioid peptides, which are neurotransmitters that act via specific opioid receptors. Among these are the two pentapeptide enkephalins. These each contain the amino acid sequence Tyr-Gly-Gly-Phe as the message domain, linked to either leucine or methionine, and are called leu-enkephalin and met-enkephalin. Other agonists incorporating the same amino acid sequence are dynorphins A and B and the most potent agonist β-endorphin, a 31-amino acid peptide with met-enkephalin at its carboxyl end. Two further peptides, endomorphins 1 and 2, have been identified that have Tyr-Pro-Phe and Tyr-Pro-Trp message domain sequences. All opioid peptides are derived by selective cleavage of larger precursor molecules.
Opioid receptors are found on the presynaptic and postsynaptic membranes of neurons in CNS pain pathways, and also in the peripheral nervous system. Three major classes of opioid receptor have been identified, which mediate distinct effects: the µ (mu), κ (kappa) and δ (delta) receptors, also termed MOP, KOP and DOP, respectively (Box 19.1). There is a distinctive regional distribution of opioid peptides and their receptors in the CNS, with high concentrations in the limbic system and spinal cord. These regions also contain high concentrations of a neutral endopeptidase (enkephalinase), which rapidly hydrolyses the pentapeptides into fragments. The various endogenous opioid peptides show preferential receptor-binding affinities: β-endorphin binds equally to µ- and δ-receptors, endomorphins bind mainly to µ-receptors, dynorphins bind preferentially to κ-receptors and the enkephalins bind preferentially to δ-receptors. The different physiological effects produced by these receptors are due to their specific neuronal distributions. An ‘orphan’ opioid receptor has been identified that does not bind the major opioid peptides, which has been named the nociceptin receptor after its specific endogenous agonist. It is widely distributed in the brain and facilitates descending enkephalinergic pathways in the spinal cord.
Opioid receptors are found on both presynaptic and postsynaptic neurons. The postsynaptic actions inhibit neuronal depolarisation, and the presynaptic effect inhibits neurotransmitter release. All opioid receptors are coupled to inhibitory G-proteins (Gi/G0), and receptor activation has many intracellular consequences:
 inhibition of adenylyl cyclase with reduced intracellular generation of cAMP, which reduces neurotransmitter release,
inhibition of adenylyl cyclase with reduced intracellular generation of cAMP, which reduces neurotransmitter release, phosphorylation of extracellular signal-regulated kinase (ERK) may be important in suppression of the immune system,
phosphorylation of extracellular signal-regulated kinase (ERK) may be important in suppression of the immune system, activation of voltage-gated inwardly rectifying K+ channels, which hyperpolarises the target cells, making them less responsive to depolarising impulses,
activation of voltage-gated inwardly rectifying K+ channels, which hyperpolarises the target cells, making them less responsive to depolarising impulses,
Morphine and synthetic opioid analgesics produce their effects largely by acting as agonists at specific opioid receptors in the CNS. Opioid drugs show receptor selectivity and can have agonist, partial agonist or antagonist properties at various opioid receptor types (Table 19.1). The analgesic action of opioids is the result of a complex series of neuronal interactions. In the nucleus raphe magnus of the brain, µ-receptor stimulation decreases activity in inhibitory GABA neurons that project to descending inhibitory serotonergic neurons in the brainstem. These neurons in turn connect presynaptically with afferent nociceptive fibres in the dorsal horn of the spinal cord. Inhibition of the GABA neurons permits increased firing of the descending inhibitory serotonergic neurons. Analgesia is produced by inhibition of the release of the pain pathway mediators, substance P, glutamate and nitric oxide from the afferent nociceptive neurons (Fig. 19.5). Activation of κ-receptors antagonises the analgesia produced by µ-receptor stimulation, by inhibiting the descending (pain-modulating) serotonergic neurons in the pain pathway. However, there is a paradoxical spinal analgesic effect from unopposed κ-receptor activation.
Opioid receptors are also present on peripheral nerves in the pain pathways, and a µ-receptor agonist reduces the sensitivity of peripheral nociceptive neurons to pain stimuli, particularly in inflamed tissues. Non-neuronal κ-receptors are involved in the inflammatory response, and are found on endothelial cells, T-lymphocytes and macrophages; κ-receptor agonists are being developed to modulate the inflammatory response orchestrated by these cells.
 Full agonists: these act principally at µ-receptors and include morphine, diamorphine, fentanyl, pethidine, codeine and dihydrocodeine. They also have weak agonist activity at δ- and κ-receptors.
Full agonists: these act principally at µ-receptors and include morphine, diamorphine, fentanyl, pethidine, codeine and dihydrocodeine. They also have weak agonist activity at δ- and κ-receptors. Mixed agonist–antagonist: pentazocine has agonist effects at the κ-receptor (and, to a lesser extent, the δ-receptor) and is a weak µ-receptor antagonist.
Mixed agonist–antagonist: pentazocine has agonist effects at the κ-receptor (and, to a lesser extent, the δ-receptor) and is a weak µ-receptor antagonist.
Some opioids have additional properties: meptazinol is a µ-receptor agonist with muscarinic receptor agonist activity; tramadol and methadone are µ-receptor agonists that also inhibit neuronal noradrenaline and serotonin uptake. These supplementary actions of tramadol and methadone contribute to their analgesic actions, since amine-mediated neurotransmission potentiates descending inhibitory pain pathways (Fig. 19.4). Methadone is also an antagonist at glutamate N-methyl-D-aspartate (NMDA) receptors, an action which can also inhibit pain transmission (Fig. 19.5).
Opioid receptor antagonists such as naloxone have no analgesic actions and are used in the treatment of opioid overdose (Ch. 53).
Effects and clinical uses
Effects on the central nervous system:
Analgesia: The analgesia produced by morphine is most effective for chronic visceral pain, but can still be helpful for some types of neuropathic pain. In addition to its antinociceptive effect, morphine alters the perception of pain, making it less unpleasant. This supraspinal effect, possibly at the limbic system, is less marked with some opioids such as pentazocine. Opioid analgesics have no anti-inflammatory effect. In fact, morphine can release the inflammatory mediator histamine locally at the site of an injection. Full µ-receptor agonists are the most powerful opioid analgesics (see Table 19.1 for list of full and partial agonists). However, some full µ-receptor agonists, for example codeine, have a low affinity for the receptors and have a limited analgesic effect. There is growing evidence that the antagonist action of methadone at NMDA receptors can produce effective analgesia in people who have become tolerant to high doses of morphine (see below). Short-acting opioids such as fentanyl and remifentanil are used for analgesia during general anaesthesia (Ch. 17).
The ceiling analgesic effect of a µ-receptor partial agonist is lower than that of a full agonist. If a person receiving high doses of a potent full µ-receptor agonist is given a µ-receptor partial agonist (e.g. buprenorphine) or a µ-receptor antagonist (e.g. pentazocine), then some of the full agonist molecules will be displaced from receptor sites by the less effective partial agonist molecules or antagonist molecules. The degree of analgesia may then be reduced, and in dependent individuals withdrawal symptoms can be produced (see below and Table 19.1).
Euphoria: The use of morphine is often associated with an elevated sense of well-being (euphoria, mediated by µ-receptors), an action that contributes considerably to its analgesic efficacy. The opposite effect (dysphoria, mediated by agonist activity at κ-receptors) counteracts the euphoric action, and the degree of euphoria produced will depend on the receptor-binding characteristics of the drug.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


