Oligomeganephronia
Aleksandr Vasilyev, MD, PhD
Key Facts
Etiology/Pathogenesis
Reduced nephron formation during development leads to nephron hypertrophy, secondary FSGS, and eventually ESRD
Most cases sporadic, some related to mutations in PAX2, EYA1, SIX1, SIX5, or HNF-1B
Macroscopic Features
Small, dense kidneys with reduced number of renal lobes
Microscopic Pathology
Fewer than normal glomeruli
Hypertrophy of remaining glomeruli and proximal tubules
Focal interstitial fibrosis and tubular atrophy
Secondary FSGS
Top Differential Diagnoses
Nephronophthisis
Dysplasia
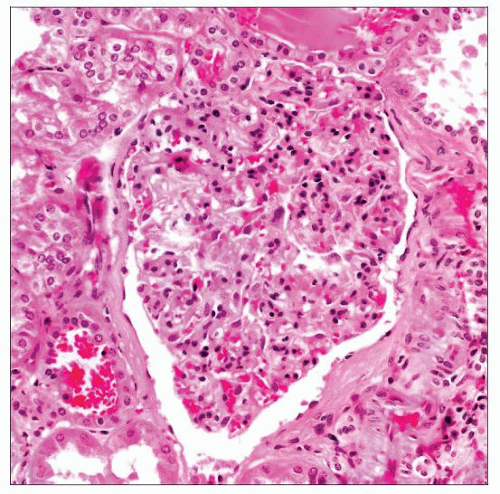 The hallmark of oligomeganephronia (OMN) is hypertrophy of the glomeruli due to a compensatory response to a congenital deficiency of nephrons. Glomeruli also have mesangial hypercellularity. |
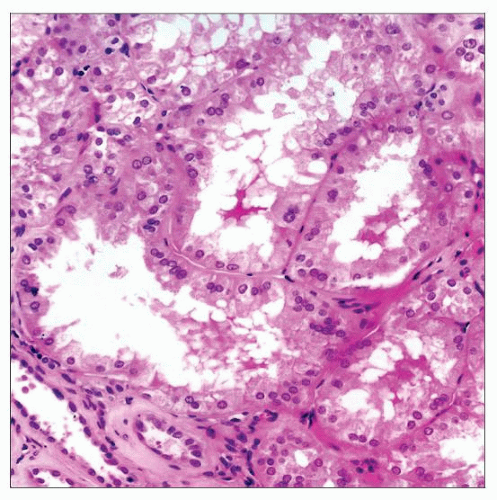 The tubules in OMN respond to congenitally decreased nephron numbers by increasing their diameter, as shown in these hypertrophied proximal tubules. |
TERMINOLOGY
Abbreviations
Oligomeganephronia (OMN)
Synonyms
Oligomeganephronic hypoplasia
Definitions
Renal hypoplasia with marked compensatory nephron hypertrophy occurring both sporadically and due to specific genetic disorders
ETIOLOGY/PATHOGENESIS
Sporadic (Most Common)
Occasional PAX2 mutations, but in most cases etiology is unknown
Prematurity and low birth weight
Genetic Disorders (Rare)
Papillorenal syndrome (OMIM 120330), a.k.a. renal-coloboma syndrome
PAX2 mutation in 50%
Associated with optic disc/nerve abnormality
Autosomal dominant, chromosome 10q
Wolf-Hirschhorn syndrome (OMIM 194190)
Monosomy 4p, multiple congenital anomalies
Branchio-oto-renal syndrome (OMIM 113650, 610896, and 600963), a.k.a. Melnick-Fraser syndrome
Brachial fistulae/clefts, ear malformations, and OMN
EYA1 (40%), SIX5, and SIX1 genes combined are involved in about 50% of all cases
Autosomal dominant
HNF-1B mutations
Acrorenal syndrome (OMIM 102520)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


