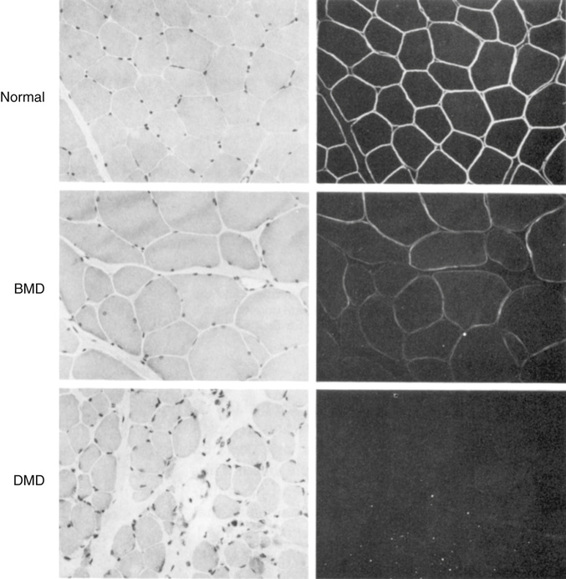
The Clinical Phenotype of Becker Muscular Dystrophy.
Becker muscular dystrophy (BMD) is also due to mutations in the dystrophin gene, but the BMD alleles produce a much milder phenotype. Patients are said to have BMD if they are still walking at the age of 16 years. There is significant variability in the progression of the disease, and some patients remain ambulatory for many years. In general, patients with BMD carry mutated alleles that maintain the reading frame of the protein and thus express some dystrophin, albeit often an altered product at reduced levels. Dystrophin is generally demonstrable in the muscle of patients with BMD (Fig. 12-17). In contrast, patients with DMD have little or no detectable dystrophin.
The Genetics of Duchenne Muscular Dystrophy and Becker Muscular Dystrophy
Inheritance.
DMD has an incidence of approximately 1 in 3300 live male births, with a calculated mutation rate of 10−4, an order of magnitude higher than the rate observed in genes involved in most other genetic diseases (see Chapter 4). In fact, given a production of approximately 8 × 107 sperm per day, a normal male produces a sperm with a new mutation in the DMD gene every 10 to 11 seconds! In Chapter 7, DMD was presented as a typical X-linked recessive disorder that is lethal in males, so that one third of cases are predicted to be due to new mutations and two thirds of patients have carrier mothers (see also Chapter 16). The great majority of carrier females have no clinical manifestations, although approximately 70% have slightly elevated levels of serum creatine kinase. In accordance with random inactivation of the X chromosome (see Chapter 6), however, the X chromosome carrying the normal DMD allele appears to be inactivated above a critical threshold of cells in some female heterozygotes. Nearly 20% of adult female carriers have some muscle weakness, whereas in 8%, life-threatening cardiomyopathy and serious proximal muscle disability occur. In rare instances, females have been described with DMD. Some have X;autosome translocations (see Chapter 6), whereas others have only one X chromosome (Turner syndrome) with a DMD mutation on that chromosome.
BMD accounts for approximately 15% of the mutations at the locus. An important genetic distinction between these allelic phenotypes is that whereas DMD is a genetic lethal, the reproductive fitness of males with BMD is high (up to approximately 70% of normal), so that they can transmit the mutant gene to their daughters. Consequently, and in contrast to DMD, a high proportion of BMD cases are inherited, and relatively few (only approximately 10%) represent new mutations.
The DMD Gene and Its Product.
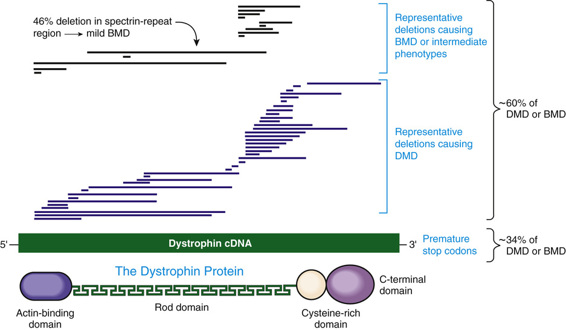
The most remarkable feature of the DMD gene is its size, estimated to be 2300 kb, or 1.5% of the entire X chromosome. This huge gene is among the largest known in any species, by an order of magnitude. The high mutation rate can be at least partly explained by the fact that the locus is a large target for mutation but, as described later, it is also structurally prone to deletion and duplication. The DMD gene is complex, with 79 exons and seven tissue-specific promoters. In muscle, the large (14-kb) dystrophin transcript encodes a huge 427-kD protein (Fig. 12-18). In accordance with the clinical phenotype, the protein is most abundant in skeletal and cardiac muscle, although many tissues express at least one dystrophin isoform.
The Molecular and Physiological Defects in Becker Muscular Dystrophy and Duchenne Muscular Dystrophy.
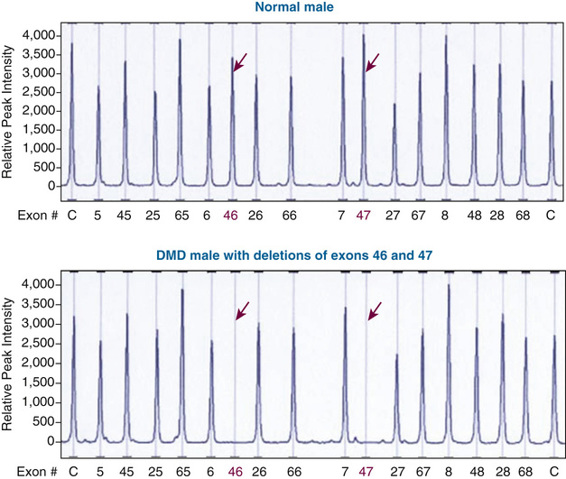
The most common molecular defects in patients with DMD are deletions (60% of alleles) (see Figs. 12-18 and 12-19), which are not randomly distributed. Rather, they are clustered in either the 5′ half of the gene or in a central region that encompasses an apparent deletion hot spot (see Fig. 12-18). The mechanism of deletion in the central region is unknown, but it appears to involve the tertiary structure of the genome and, in some cases, recombination between Alu repeat sequences (see Chapter 2) in large central introns. Point mutations account for approximately one third of the alleles and are randomly distributed throughout the gene.
The absence of dystrophin in DMD destabilizes the myofiber membrane, increasing its fragility and allowing increased Ca++ entry into the cell, with subsequent activation of inflammatory and degenerative pathways. In addition, the chronic degeneration of myofibers eventually exhausts the pool of myogenic stem cells that are normally activated to regenerate muscle. This reduced regenerative capacity eventually leads to the replacement of muscle with fat and fibrotic tissue.
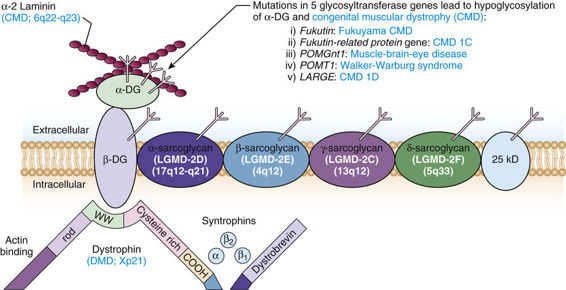
The Dystrophin Glycoprotein Complex (DGC).
Dystrophin is a structural protein that anchors the DGC at the cell membrane. The DGC is a veritable constellation of polypeptides associated with a dozen genetically distinct muscular dystrophies (Fig. 12-20). This complex serves several major functions. First, it is thought to be essential for the maintenance of muscle membrane integrity, by linking the actin cytoskeleton to the extracellular matrix. Second, it is required to position the proteins in the complex at the sarcolemma. Although the function of many of the proteins in the complex is unknown, their association with diseases of muscle indicates that they are essential components of the complex. Mutations in several of these proteins cause autosomal recessive limb girdle muscular dystrophies and other congenital muscular dystrophies (Fig. 12-20).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


