Disorders of Gonadal Development
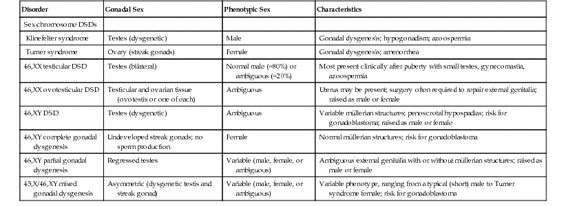
Gonadal dysgenesis refers to a progressive loss of germ cells, typically leading to underdeveloped and dysfunctional (“streak”) gonads, with consequent failure to develop mature secondary sex characteristics. Gonadal dysgenesis is typically categorized according to the karyotype of a patient. Complete gonadal dysgenesis (CGD)—as in the case of XX males (now formally designated 46,XX testicular DSD) or XY females (now formally designated 46,XY CGD)—is characterized by normal-appearing external genitalia of the opposite chromosomal sex. Cases with ambiguous external genitalia are said to have partial gonadal dysgenesis. Gonadal dysgenesis can also be associated with sex chromosome DSDs; it is a consistent feature of Turner syndrome (see Table 6-7), and patients with a 45,X/46,XY karyotype have mixed gonadal dysgenesis.
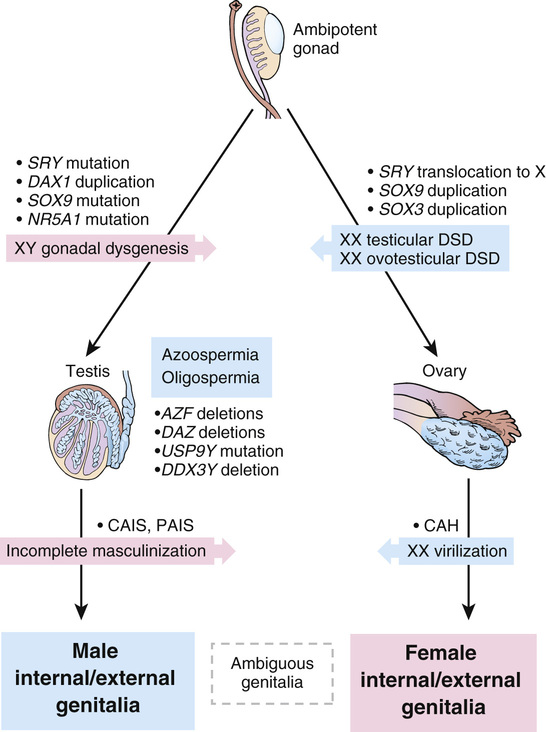
Various types of gonadal dysgenesis, their clinical phenotypes, and genetic causes are summarized in Table 6-9 and illustrated schematically in Figure 6-16.
Disorders Associated with a 46,XY Karyotype
We begin with DSDs associated with a 46,XY karyotype. The overall incidence of these conditions is approximately 1 in 20,000 live births. Although a number of cytogenetic or single-gene defects have been demonstrated, many such cases remain unexplained. Approximately 15% of patients with 46,XY CGD have deletions or mutations in the SRY gene that interfere with the normal male pathway. However, most females with a 46,XY karyotype have an apparently normal SRY gene.
The DAX1 gene in Xp21.3 encodes a transcription factor that plays a dosage-sensitive role in determination of gonadal sex, implying a tightly regulated interaction between DAX1 and SRY. Although production of SRY at a critical point in early development normally leads to testis formation, an excess of DAX1 resulting from duplication of the gene can apparently suppress the normal male-determining function of SRY, leading to ovarian development (see Fig. 6-16).
A key master gene in gonadal development and the target of SRY signaling is the SOX9 gene on chromosome 17. SOX9 is normally expressed early in development in the genital ridge and is required for normal testis formation. Mutations in one copy of the SOX9 gene, typically associated with a skeletal malformation disorder called camptomelic dysplasia, lead to complete gonadal dysgenesis in approximately 75% of 46,XY cases (see Table 6-8). In the absence of one copy of the SOX9 gene, testes fail to form, and the ovarian pathway is followed instead. The phenotype of these patients suggests that the critical step for the male pathway is sufficient SOX9 expression to drive the formation of testes, normally after up-regulation by the SRY gene. In 46,XY CGD, with either a mutation in SRY or a mutation in SOX9, the levels of SOX9 expression remain too low for testis differentiation, allowing ovarian differentiation to ensue.
As many as 10% of patients with a range of 46,XY DSD phenotypes carry mutations in the NR5A1 gene, which encodes a transcriptional regulator of a number of genes, including SOX9 and DAX1. These mutations are associated with inadequate androgenization of external genitalia, leading to ambiguous genitalia, partial gonadal dysgenesis, and absent or rudimentary müllerian structures.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


