A multigenic model has also been observed in a few families with Bardet-Biedl syndrome, a rare birth defect characterized by obesity, variable degrees of intellectual disability, retinal degeneration, polydactyly, and genitourinary malformations. Fourteen different genes have been found in which mutations cause the syndrome. Although inheritance is clearly autosomal recessive in most families, a few families appear to demonstrate digenic inheritance, in which the disease occurs only when an individual is homozygous for mutations at one of these 14 loci and is heterozygous for a mutation at another of the loci.
Gene-Environment Interactions in Venous Thrombosis
Another example of gene-gene interaction predisposing to disease is found in the group of conditions referred to as hypercoagulability states, in which venous or arterial clots form inappropriately and cause life-threatening complications of thrombophilia (Case 46). With hypercoagulability, however, there is a third factor, an environmental influence that in the presence of the predisposing genetic factors, increases the risk for disease even more.
One such disorder is idiopathic cerebral vein thrombosis, a disease in which clots form in the venous system of the brain, causing catastrophic occlusion of cerebral veins in the absence of an inciting event such as infection or tumor. It affects young adults, and although quite rare (<1 per 100,000 in the population), it carries a high mortality rate (5% to 30%). Three relatively common factors—two genetic and one environmental—that lead to abnormal coagulability of the clotting system are each known to individually increase the risk for cerebral vein thrombosis (Fig. 8-8):
• A missense variant in the gene for the clotting factor, factor V
• A variant in the 3′ untranslated region (UTR) of the gene for the clotting factor prothrombin
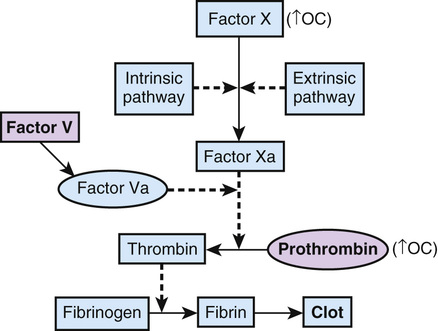
A polymorphic allele of factor V, factor V Leiden (FVL), in which arginine is replaced by glutamine at position 506 (Arg506Gln), has a frequency of approximately 2.5% in white populations but is rarer in other population groups. This alteration affects a cleavage site used to degrade factor V, thereby making the protein more stable and able to exert its procoagulant effect for a longer duration. Heterozygous carriers of FVL, approximately 5% of the white population, have a risk for cerebral vein thrombosis that, although still quite low, is sevenfold higher than that in the general population; homozygotes have a risk that is eightyfold higher.
The second genetic risk factor, a mutation in the prothrombin gene, changes a G to an A at position 20210 in the 3′ UTR of the gene (prothrombin g.20210G>A). Approximately 2.4% of white individuals are heterozygotes, but it is rare in other ethnic groups. This change appears to increase the level of prothrombin mRNA, resulting in increased translation and elevated levels of the protein. Being heterozygous for the prothrombin 20210G>A allele raises the risk for cerebral vein thrombosis three to sixfold.
Finally, the use of oral contraceptives containing synthetic estrogen increases the risk for thrombosis fourteen- to twentytwofold, independent of genotype at the factor V and prothrombin loci, probably by increasing the levels of many clotting factors in the blood. Although using oral contraceptives and being heterozygous for FVL cause only a modest increase in risk compared with either factor alone, oral contraceptive use in a heterozygote for prothrombin 20210G>A raises the relative risk for cerebral vein thrombosis 30- to 150-fold!
There is also interest in the role of FVL and prothrombin 20210G>A alleles in deep venous thrombosis (DVT) of the lower extremities, a condition that occurs in approximately 1 in 1000 individuals per year, far more common than idiopathic cerebral venous thrombosis. Mortality due to DVT (primarily due to pulmonary embolus) can be up to 10%, depending on age and the presence of other medical conditions. Many environmental factors are known to increase the risk for DVT and include trauma, surgery (particularly orthopedic surgery), malignant disease, prolonged periods of immobility, oral contraceptive use, and advanced age.
The FVL allele increases the relative risk for a first episode of DVT sevenfold in heterozygotes; heterozygotes who use oral contraceptives see their risk increased thirtyfold compared with controls. Heterozygotes for prothrombin 20210G>A also have an increase in their relative risk for DVT of twofold to threefold. Notably, double heterozygotes for FVL and prothrombin 20210G>A have a relative increased risk of twentyfold—a risk approaching a few percent of the population.
Thus each of these three factors, two genetic and one environmental, on its own increases the risk for an abnormal hypercoagulable state; having two or all three of these factors at the same time raises the risk even more, to the point that thrombophilia screening programs for selected populations of patients may be indicated in the future.
Multiple Coding and Noncoding Elements in Hirschsprung Disease
A more complicated set of interacting genetic factors has been described in the pathogenesis of a developmental abnormality of the enteric nervous system in the gut known as Hirschsprung disease (HSCR) (Case 22). In HSCR, there is complete absence of some or all of the intrinsic ganglion cells in the myenteric and submucosal plexuses of the colon. An aganglionic colon is incapable of peristalsis, resulting in severe constipation, symptoms of intestinal obstruction, and massive dilatation of the colon (megacolon) proximal to the aganglionic segment. The disorder affects approximately 1 in 5000 newborns of European ancestry but is twice as common among Asian infants. HSCR occurs as an isolated birth defect 70% of the time, as part of a chromosomal syndrome 12% of the time, and as one element of a broad constellation of congenital abnormalities in the remainder of cases. Among patients with HSCR as an isolated birth defect, 80% have only a single, short aganglionic segment of colon at the level of the rectum (hence, HSCR-S), whereas 20% have aganglionosis of a long segment of colon, the entire colon or, occasionally, the entire colon plus the ileum as well (hence, HSCR-L).
Familial HSCR-L is often characterized by patterns of inheritance that suggest dominant or recessive inheritance, but consistently with reduced penetrance. HSCR-L is most commonly caused by loss-of-function missense or nonsense mutations in the RET gene, which encodes RET, a receptor tyrosine kinase. A small minority of families have mutations in genes encoding ligands that bind to RET, but with even lower penetrance than those families with RET mutations.
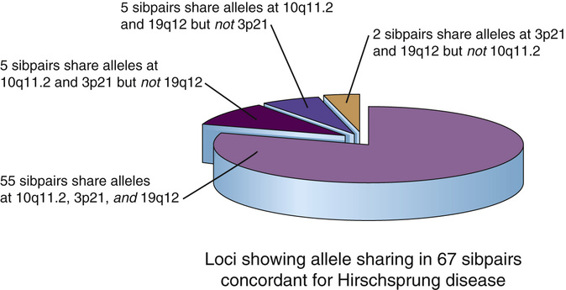
HSCR-S is the more common type of HSCR and has many of the characteristics of a disorder with complex genetics. The relative risk ratio for sibs, λs, is very high (approximately 200), but MZ twins do not show perfect concordance and families do not show any obvious mendelian inheritance pattern for the disorder. When pairs of siblings concordant for HSCR-S were analyzed genome-wide to see which loci and which sets of alleles at these loci each sib had in common with an affected brother or sister, alleles at three loci (including RET) were found to be significantly shared, suggesting gene-gene interactions and/or multigenic inheritance; indeed, most of the concordant sibpairs were found to share alleles at all three loci. Although the non-RET loci have yet to be identified, Figure 8-9 illustrates the range of interactions necessary to account for much of the penetrance of HSCR in even this small cohort of patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


