1. Normal glomeruli by light microscopy
(a) Minimal change disease
(b) Thin glomerular basement membrane disease
(c) Early Alport disease
(d) Early membranous glomerulonephritis
2. Focal/segmental lesions
(a) Focal segmental glomerulosclerosis
• Idiopathic vs. secondary
(b) Focal segmental proliferative glomerulonephritis
(c) Focal segmental necrotizing and crescentic glomerulonephritis
3. Diffuse glomerulonephritis
(a) Membranous glomerulonephritis
• Idiopathic vs. secondary
(b) Proliferative glomerulonephritis
• Mesangial proliferative glomerulonephritis
– IgA nephropathy
– Mesangial proliferative lupus nephritis
• Endocapillary proliferative glomerulonephritis
– Acute postinfectious glomerulonephritis
• Mesangiocapillary glomerulonephritis
– C3 glomerulonephritis
– Membranoproliferative glomerulonephritis (MPGN)
– Proliferative glomerulonephritis with monoclonal IgG deposits
– Diffuse proliferative lupus nephritis
– Cryoglobulinemic glomerulonephritis
– Chronic thrombotic microangiopathy
4. Crescentic glomerulonephritis
(a) Immune complex-mediated crescentic glomerulonephritis
(b) Pauci-immune (ANCA associated) crescentic glomerulonephritis
(c) Antiglomerular basement membrane antibody mediated
5. Diffuse/nodular mesangial expansion
(a) Diabetic glomerulosclerosis
(b) Light chain deposition disease
(c) Fibrillary/Immunotactoid glomerulonephritis
(d) Amyloidosis
Primary Glomerular Diseases
Minimal Change Disease (MCD)
Clinical
♦
The most common cause of the nephrotic syndrome in children (Table 34.2)
Table 34.2.
Classification of Primary Glomerulonephritis by Predominant Clinical Manifestations
Nephrotic syndrome | Nephritic syndrome | |
|---|---|---|
Minimal change disease | +++++ | – |
Focal segmental | ||
Glomerulosclerosis | ++++ | + |
Membranous | ||
Glomerulonephritis | ++++ | + |
IgA nephropathy | +++ | ++ |
C3Glomerulonephritis | +++ | +++ |
Proliferative glomerulonephritis with monoclonal IgG deposits | +++ | +++ |
Membranoproliferative GN | ++ | +++ |
Acute postinfectious GN | + | ++++ |
Crescentic | ||
Glomerulonephritis | + | ++++ |
♦
Mild periorbital edema, prior to the rapid onset of the nephrotic syndrome
♦
Proteinuria is “selective” or composed primarily of albumin
♦
Microscopic hematuria is rare; hypertension is unusual
♦
MCD in adults can be associated with various drugs (i.e., NSAIDs)
♦
The pathogenesis has been related to abnormalities in ANGPTL4 expression in some cases
Microscopic
♦
Light microscopy
The glomeruli, tubules, and interstitium appear normal (Fig. 34.1).
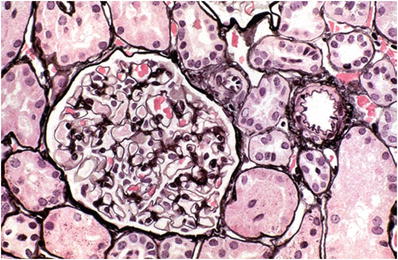
Fig. 34.1.
Minimal change disease with normal glomerulus, tubules, and arteriole.
♦
Immunofluorescence microscopy
Usually negative, mesangial IgM may occasionally be present (“IgM nephropathy” in cases with >2+ staining).
Differential Diagnosis
♦
An early membranous lesion may look normal in light microscopy
♦
Undersampled focal segmental glomerulosclerosis (where segmental scars are not present in the biopsy) may look identical to MCD
Focal Segmental Glomerulosclerosis (FSGS)
Clinical
♦
Focal segmental glomerulosclerosis may be primary (idiopathic) or secondary to a number of etiologic agents, including the following:
Unilateral renal agenesis
Renal ablation
Sickle cell disease
Morbid obesity (with or without sleep apnea)
Reflux nephropathy
HIV nephropathy
♦
Idiopathic FSGS is the most common cause of nephrotic syndrome in African Americans
A subset of cases is associated with elevated suPAR levels.
Childhood onset of FSGS has been linked to podocin gene mutations
♦
Secondary FSGS usually does not present with the full nephrotic syndrome
Microscopic
♦
Light microscopy
Focal and segmental glomerular sclerosis with capillary loop collapse, hyaline and lipid deposition, and often adhesion to Bowman capsule (Fig. 34.3)
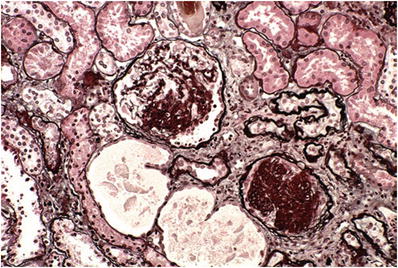
Fig. 34.3.
Segmental (upper glomerulus) and global glomerulosclerosis (lower glomerulus) on silver stain.
The remainder of the glomerular tuft appears normal
Lesions begin or are more common near the corticomedullary junction.
♦
Immunofluorescence microscopy
May be negative
Deposition of IgM and C3 in mesangium or in segmental sclerosis
♦
Electron microscopy (also see Chapter 4)
Effacement of podocyte foot processes
Podocyte denudation may be present focally as an early lesion
Segmental sclerosis may also be seen
Differential Diagnosis
♦
May be secondary (see earlier) or the end result of segmental necrotizing diseases
Membranous Nephropathy (MN)
Clinical
♦
MN is the major cause of nephrotic syndrome in adults
♦
It may be primary or occur in association with a number of disorders or exposure to antigenic substances (“secondary MGN” which accounts for up to 30% of cases), such as:
Systemic lupus erythematosus
Malignant neoplasms
Exposure to gold and mercury
Penicillamine, captopril, and NSAIDS
Hepatitis B
Various metabolic disorders
♦
MN may be a disease in which autoantigens are planted within the subepithelial space of the glomerular capillary loops or associated with autoanti-PLA2 antibodies, the target of which is expressed on podocytes
♦
Immunofluorescence staining for PLA2R is useful in distinguishing idiopathic from secondary MN and recurrent from de novo MN in the post-renal transplant setting
Microscopic
♦
Light microscopy (Fig. 34.4)
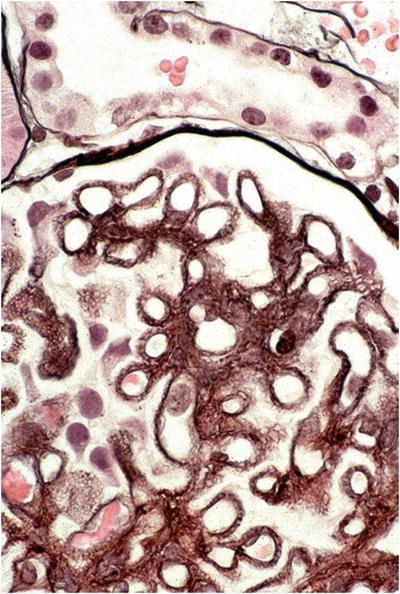
Fig. 34.4.
Membranous nephropathy with subepithelial basement membrane “spikes” on silver stain.
The glomeruli may appear normal if the deposits are small (early disease)
Capillary walls are thickened, with subepithelial spikes on silver stain
♦
Immunofluorescence microscopy
Bright granular staining of the capillar y loops with anti-IgG and C3 (Fig. 34.5)
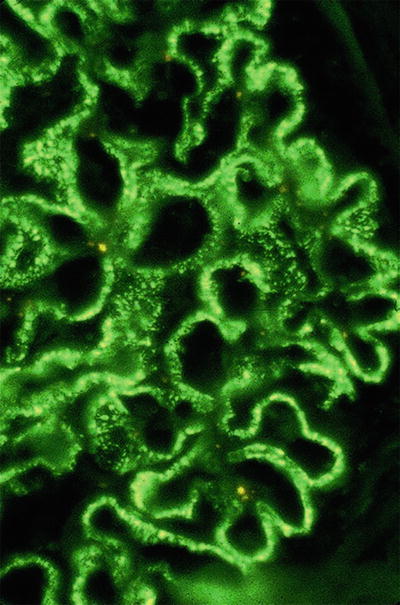
Fig. 34.5.
Granular capillary wall staining with anti-IgG in membranous nephropathy.
♦
Electron microscopy (also see Chapter 4)
Subepithelial electron-dense deposits with intervening basement membrane spikes (Fig. 34.6)
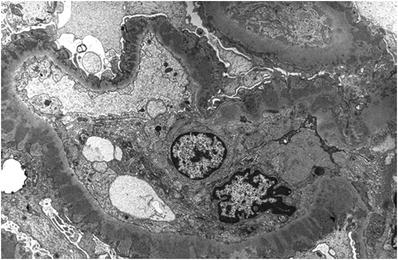
Fig. 34.6.
Electron micrograph of subepithelial electron-dense deposits and adjacent GBM spikes in membranous nephropathy.
Stage of disease correlates with incorporation of deposits into the glomerular basement membrane (GBM)
Differential Diagnosis
♦
As noted above, secondary causes of MGN are common (especially systemic lupus erythematosus)
In lupus, mesangial deposits and tubuloreticular inclusions and staining with C1q are seen.
♦
An early lesion may be confused with minimal change disease (MCD) by light microscopy
IgA Nephropathy (IgAN)
Clinical
♦
Most common form of primary glomerulonephritis in the world
♦
IgAN has a higher incidence in patients of Asian and Native American descent
♦
It may be related to a genetic or acquired abnormality of immune regulation leading to increased mucosal IgA synthesis in response to respiratory or gastrointestinal exposure to environmental agents
♦
Occurs with increased frequency in patients with:
Celiac disease
Dermatitis herpetiformis
Liver disease
♦
Patients usually present with one of three syndromes:
Macroscopic hematuria concurrent with an upper respiratory infection, the so-called synpharyngitic hematuria
Asymptomatic microscopic hematuria and variable proteinuria, including the nephrotic syndrome
Henoch–Schonlein purpura is the systemic form of the disease process causing IgA nephropathy and occurs more frequently in children than adults
•
Patients with Henoch–Schonlein purpura manifest skin, joint, and intestinal involvement
Microscopic
♦
Light microscopy
The glomeruli show varying degrees of mesangial hypercellularity (Fig. 34.7)
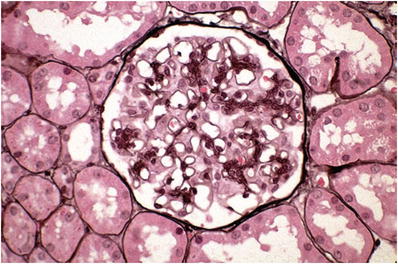
Fig. 34.7.
Glomerulus with mild mesangial proliferation in IgA nephropathy.
Segmental proliferation, segmental sclerosis, and necrosis with crescents may be seen
♦
Immunofluorescence microscopy
Mesangial deposits of IgA (dominant or codominant); IgG and C3 are variably present (Fig. 34.8)
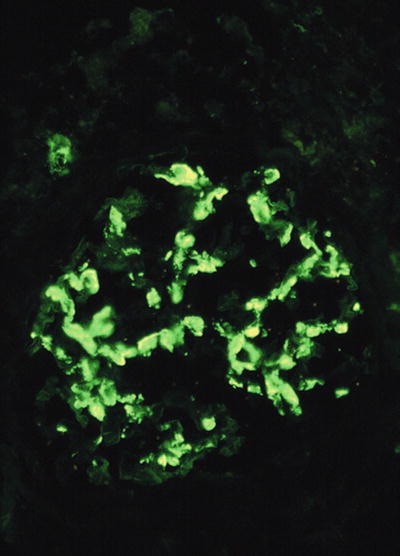
Fig. 34.8.
Granular mesangial staining with anti-IgA in IgA nephropathy.
♦
Electron microscopy
Deposits are present in the mesangium often in a “paramesangial” location (beneath the basement membrane as it covers mesangium) (Fig. 34.9)
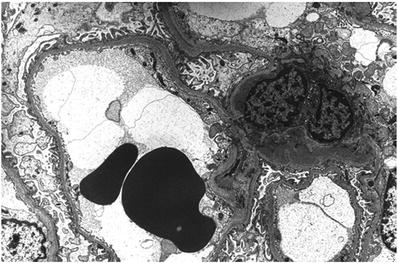
Fig. 34.9.
Paramesangial electron-dense deposits in IgA nephropathy.
Differential Diagnosis
♦
Mesangial deposits may also be seen in SLE (WHO class II lupus nephritis, usually with “full house” immunofluorescence staining) resolving postinfectious glomerulonephritis
C3 Glomerulopathy (C3G)
Clinical
♦
C3G describes conditions caused by abnormal control of the alternate complement pathway, glomerular deposition of C3, and associated electron-dense deposits by electron microscopy
♦
C3G is broadly divided into two main categories:
Dense deposit disease (DDD , formerly membranoproliferative glomerulonephritis type II ) is associated with autoantibodies to C3 convertase (“C3 nephritic factor”)
C3 glomerulonephritis (C3GN )
♦
The disease most often affects young Caucasians; C3GN has a median age of onset of 25 years and DDD 15 years
♦
Patients may present with a range of proteinuria (minimal to nephrotic range) and/or hematuria (microscopic to gross)
♦
Diagnosis of these entities should prompt investigation for abnormalities in complement factors/regulation
Microscopic
♦
Light microscopy
Much like membranoproliferative glomerulonephritis, biopsies often demonstrate mesangial hypercellularity, matrix expansion, and mesangial interposition beneath the endothelium with formation of double contours by silver staining
♦
Immunofluorescence microscopy
By definition, both forms of C3N tend to only show staining for C3 with little to no immunoglobulin
♦
Electron microscopy
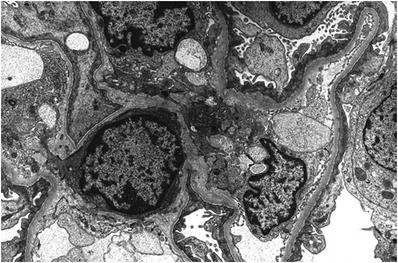
In DDD, there are “sausage-like” electron-dense deposits within lamina densa of the GBM corresponding to the C3 linear staining in the peripheral capillary loops with ring-like patterns in the mesangium (Fig. 34.10)
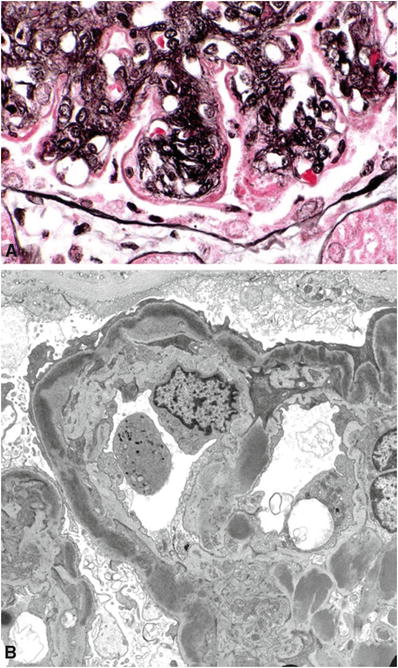
Fig. 34.10.
Dense deposit disease (MPGN type II) with thickened, silver-negative capillary wall (A). Electron micrograph of interrupted, intramembranous electron-dense deposits in dense deposit disease (B).
Cases with isolated C3 staining and electron-dense deposits that do not have the typical morphology of DDD are usually classified as C3GN
Differential Diagnosis
♦
Membranoproliferative glomerulonephritis type I may have a similar light microscopic appearance but will often have concomitant staining with immunoglobulins by immunofluorescence
♦
Distinction from resolving postinfectious glomerulonephritis can be difficult (see below). Clinical/serologic data (i.e., positive antistreptolysin O) can be helpful
Membranoproliferative (Mesangiocapillary) Glomerulonephritis (MPGN)
Clinical
♦
Chronic progressive glomerulonephritis in older children and adults
♦
Circulating immune complexes have been identified in 50% of patients
♦
Activation of the complement system with hypocomplementemia is a hallmark of MPGN
♦
Patients may present with the following:
Nephrotic syndrome
Abnormal urinary sediment with nonnephrotic proteinuria
Acute nephritis
♦
MPGN may be primary or associated with other systemic disorders (secondary), including the following:
Hepatitis B and hepatitis C infection
Infected ventriculoatrial shunts
Subacute bacterial endocarditis
Schistosomiasis
Alpha 1-antitrypsin deficiency
Chronic liver disease
♦
Three types of MPGN (types I–III) have been described based on morphology and clinical features
Microscopic
♦
The condition typically features mesangial hypercellularity, matrix expansion, and mesangial interposition beneath the endothelium with formation of double contours
Type I MPGN (Fig. 34.11)
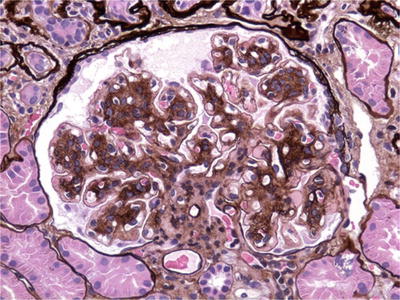
Fig. 34.11.
Mesangial and endocapillary hypercellularity with GBM duplication in MPGN type I.
•
Subendothelial and mesangial deposits that frequently contain C3 and immunoglobulins
•
Most cases of secondary MPGN are type I
Dense deposit disease was formerly known as MPGN type II (see above)
Type III MPGN
•
Prominent mesangial, subendothelial, and subepithelial immune deposits
•
Type III is the least common form of MPGN and may actually represent a form of C3 glomerulonephritis
Proliferative Glomerulonephritis with Monoclonal IgG Deposits
Clinical
♦
Similar to MPGN, patients may present with variable amounts of hematuria and/or proteinuria
♦
Only 30% of cases are associated with an underlying dysproteinemia (amyloidosis, multiple myeloma, or paraprotein)
♦
Outcome with therapy ranges from remission to end-stage renal disease, with prognosis most closely associated with the degree of glomerulosclerosis/tubular atrophy
Microscopic
♦
Light microscopy
Biopsies often demonstrate heterogeneous mesangial hypercellularity, matrix expansion, and mesangial interposition beneath the endothelium with formation of double contours by silver staining akin to MPGN
Some cases may also show “exudative” features like in postinfectious glomerulonephritis (see below), solely mesangial proliferation, or crescents
♦
Immunofluorescence microscopy
Staining is only observed in glomeruli and parallels the sites of immune complex deposition by electron microscopy
Most cases demonstrate IgG and C3 staining with κ or λ restriction
C1q may also be present
♦
Electron microscopy
Immune deposits are almost always seen in subendothelial and mesangial locations, typically global in distribution, and usually lack substructure
Subepithelial and intramembranous deposits may be identified
Differential Diagnosis
♦
Other glomerulonephritides with immune complex deposition may mimic the light microscopic appearance (particularly MPGN type I), although staining in these entities is positive for both kappa and lambda light chains
Postinfectious Glomerulonephritis (PIGN)
Clinical
♦
May be caused by a number of infectious agents
♦
Poststreptococcal glomerulonephritis is primarily a disease of children, 6–7 years of age
The onset is usually abrupt, with a latent period of 7–21 days between infection and the development of nephritis, and may result from pharyngitis (“postpharyngitic hematuria”) or skin infections
During epidemics, the clinical attack rate is 10–12%, but subclinical disease occurs four times more frequently than overt disease; asymptomatic contacts may have hematuria
♦
Common initial clinical manifestations of poststreptococcal glomerulonephritis are:
Hematuria (microscopic or macroscopic)
Edema
Hypertension
Oliguria
♦
The acute clinical episode of poststreptococcal glomerulonephritis is usually self-limited, and complement levels, which are usually depressed acutely, return to normal within 6 weeks
♦
In most patients, hematuria disappears by 6 months, but proteinuria may persist for 2 years in one-third of patients
Microscopic
♦
Light microscopy
The glomeruli show diffuse mesangial proliferation and “exudative” endocapillary hypercellularity with infiltration of neutrophils and mononuclear inflammatory cells (Fig. 34.12)
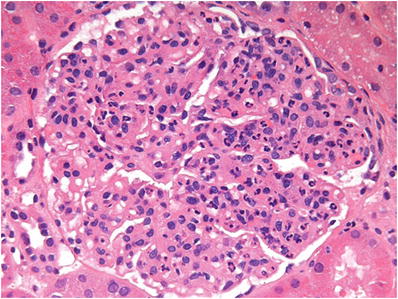
Fig. 34.12.
Diffuse mesangial and endocapillary hypercellularity in acute postinfectious glomerulonephritis.
Crescents may also be present
♦
Immunofluorescence microscopy
Granular deposits of predominantly C3 with lesser amounts of IgG along the capillary loops and in the mesangium (Fig. 34.13)
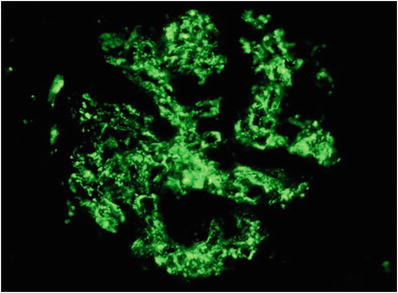
Fig. 34.13.
Granular capillary wall and mesangial staining with anti-IgG in acute postinfectious glomerulonephritis.
IgA-containing deposits may be noted in cases related to methicillin-resistant Staphylococcus aureus infections
♦
Electron microscopy
Large subepithelial, “hump-like” deposits as well as mesangial deposits (Fig. 34.14)
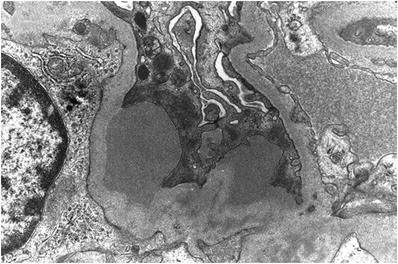
Fig. 34.14.
Large subepithelial deposits in acute postinfectious glomerulonephritis.
The capillary loop deposits become less frequent after a few weeks, but the mesangial deposits persist for a longer period
Crescentic Glomerulonephritis
Clinical
♦
May be
Immune complex mediated (any disease with immune complex deposition, such as SLE, IgAN, PIGN, etc.)
Pauci-immune (ANCA associated) accounts for approximately 50% of cases
Mediated by antiglomerular basement membrane antibody
♦
Patients with crescentic glomerulonephritis secondary to antiglomerular basement membrane disease may also have pulmonary hemorrhage (Goodpasture disease)
♦
Most cases of pauci-immune glomerulonephritis are associated with small vessel vasculitides (see later)
♦
The prognosis depends on the number of crescents present in the biopsy; a more diffuse crescentic process predicts a worse prognosis
Microscopic
♦
Light microscopy
Disruption of the glomerular capillary loops causes extravasation of the blood and cells into Bowman space, which incites proliferation of parietal epithelial cells, forming a cellular crescent (Fig. 34.15)
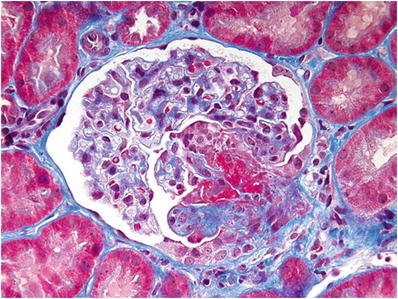
Fig. 34.15.
Segmental glomerular necrotizing lesion (lower right) with overlying cellular crescent.
♦
Immunofluorescence microscopy
Fibrinogen staining is seen in areas undergoing necrosis and crescent formation
Bright linear staining of GBM for IgG in antiglomerular basement membrane-mediated disease (Fig. 34.16)
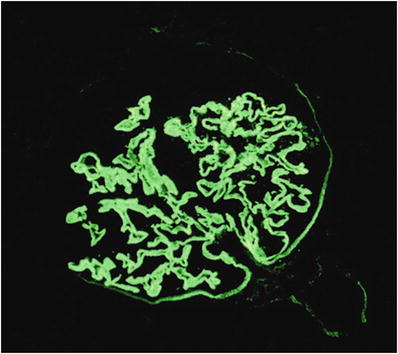
Fig. 34.16.
Bright, linear capillary wall staining with anti-IgG in anti-GBM-mediated glomerulonephritis.
Negative in pauci-immune crescentic glomerulonephritis
Other immune complexes are present in immune complex-mediated disease
♦
Electron microscopy
Disruption of capillary wall with or without immune deposits
Fibrillary Glomerulonephritis
Clinical
♦
Most patients are 40–50 years of age, with a slight female predominance and a greater frequency in whites
♦
Most patients present with proteinuria, and occasionally the nephrotic syndrome, or with symptoms of nephritis
♦
Renal insufficiency is progressive, with renal failure usually occurring within 2–4 years
Microscopic
♦
Light microscopy
The glomeruli show mesangial matrix expansion with PAS- and silver-negative material, variable hypercellularity, and capillary wall thickening (Fig. 34.17)
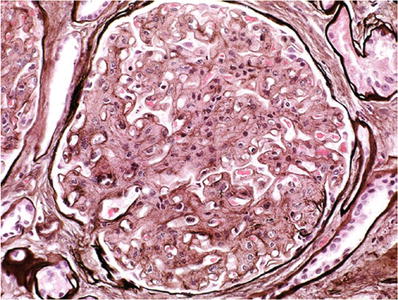
Fig. 34.17.
Fibrillary glomerulonephritis with pale mesangial expansion and thickened capillary walls on silver stain.
♦
Immunofluorescence microscope
“Smudgy” mesangial and capillary wall staining for IgG and C3 are present
If IgG subtyping is performed, IgG4 is predominant
♦
Electron microscopy
There is glomerular deposition of fibrils that are thicker than amyloid fibrils and lack Congo red birefringence (Fig. 34.18)
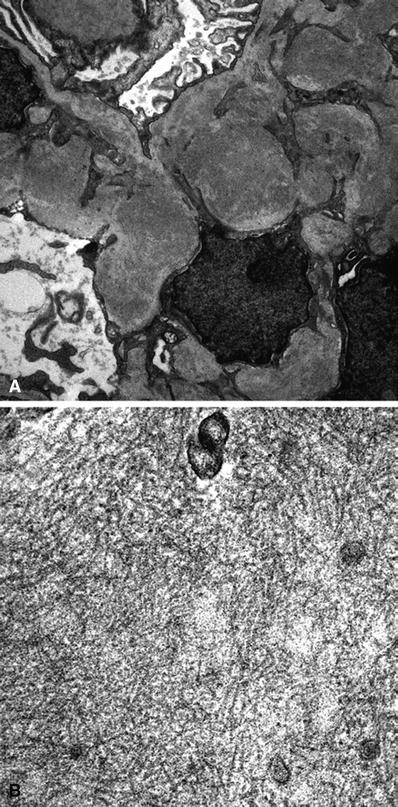
Fig. 34.18
Electron micrograph of fibrillary glomerulonephritis with mesangial electron-dense deposits at low magnification (A). Electron micrograph of fibrillary glomerulonephritis with mesangial fibrils at higher magnification (B).
The 18–22 nm randomly oriented, nonbranching fibrils are present in the mesangium and at least segmentally along the capillary loop basement membranes
Differential Diagnosis
♦
Immunotactoid glomerulopathy also presents with light microscopic features but is more often associated with a lymphoplasmacytic malignancy and has larger fibrils (>30 nm) arranged in parallel arrays
Secondary Glomerular Diseases
Diabetes Mellitus (Diabetic Nephropathy)
Clinical
♦
Major cause of end-stage renal failure in the United States
♦
Approximately one-third of patients entering dialysis programs have lost renal function as a result of diabetes
♦
Patients with diabetes mellitus rarely develop clinically detectable glomerular injury before 10 years
♦
Nodular mesangial sclerosis (Kimmelstiel–Wilson nodule) is the quintessential lesion
Microscopic
♦
The histologic changes in type 1 and type 2 diabetes are identical
♦
Light microscopy
Thickened tubular and glomerular basement (after 2–3 years)
Interstitial fibrosis and expansion of mesangial regions with formation of PAS- and silver-positive mesangial nodules begin after 3–5 years; it parallels progressive loss of renal function (Fig. 34.19)
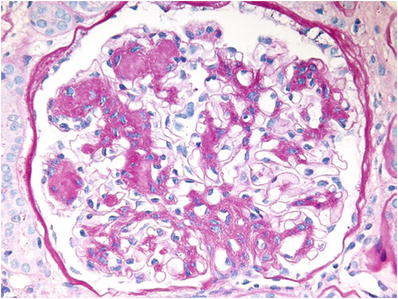
Fig. 34.19.
Diabetic glomerulosclerosis with PAS-positive mesangial nodules.
♦
Electron microscopy
Diffuse thickening of the capillary loop basement membranes and mesangial matrix expansion (Fig. 34.20)
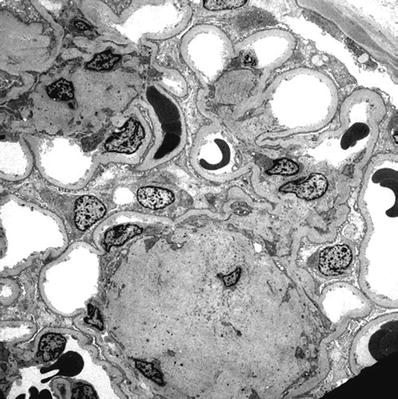
Fig. 34.20.
Electron micrograph of diabetic glomerulosclerosis with nodular mesangial sclerosis and thickened GBM.
Differential Diagnosis
♦
Both light chain deposition disease and amyloidosis may form mesangial nodules
Nodules are generally PAS negative, and immunofluorescence is positive for the corresponding immunoglobulin
Paraprotein-related Conditions
Paraproteinemia
♦
A group of disorders characterized by tissue damage associated with the overproduction of monoclonal immunoglobulin proteins and their components
♦
The renal manifestations occur as a result of the interaction of the abnormal protein with normal tissue components
Amyloidosis
♦
Extracellular deposition of insoluble fibrillar proteins with a characteristic β-pleated sheet configuration; most proteins have an α-helical structure
♦
There are at least ten proteins associated with human systemic amyloidosis: immunoglobulin light chain (AL), amyloid A (AA), transthyretin, cystatin C, apolipoprotein A-1, gelsolin, fibrinogen Aα-chain, lysozyme, apoprotein A-II, and LECT2
♦
The main types of amyloid that affect the kidney include:
Primary or AL amyloid is caused by a plasma cell dyscrasia with overproduction of a monoclonal immunoglobulin light chain, which in most patients is one light chain (usually lambda); overt myeloma is present in 20% of these patients
Secondary or AA amyloid occurs in chronic infections and chronic inflammatory states, including rheumatoid arthritis, ankylosing spondylitis, tuberculosis, osteomyelitis, and intravenous drug abuse
LECT2-associated amyloidosis, first described in 2008, is characterized by extensive renal glomerular, vascular, and interstitial amyloid deposition as well as liver involvement. Individuals of Mexican heritage are commonly affected
Microscopic
♦
Light microscopy
Amyloid deposits occur predominantly in glomeruli and appear as amorphous, eosinophilic nodules within the mesangium and as thickened capillaries
The amyloid deposits do not stain with the silver stain (nonargyrophilic), are PAS weak or negative, and show green birefringence under polarized light with Congo red staining (Fig. 34.21)
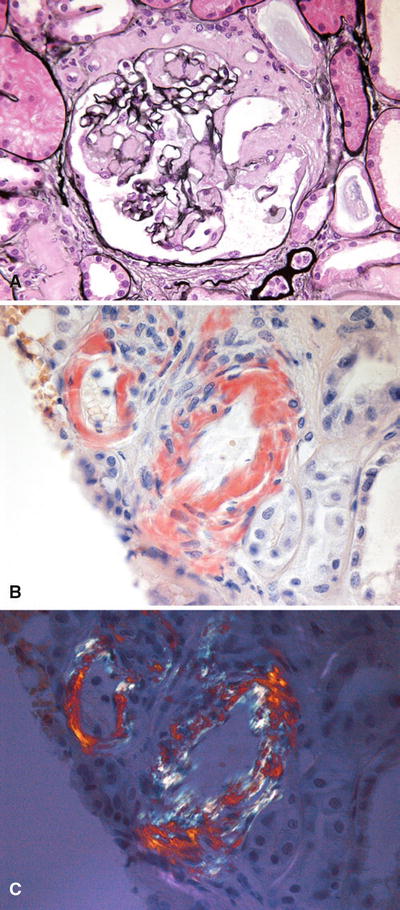
Fig. 34.21.
Pale-staining glomerular amyloid deposition on silver stain (A). Arteriolar amyloid showing Congo red positivity (B). Arteriolar amyloid showing positive birefringence of Congo red stain with polarized light (C).
♦
Immunofluorescence microscopy
Amyloid deposits of AL type show staining restricted to one light chain (usually lambda)
♦
Electron microscopy
The amyloid deposits are composed of nonbranching, randomly oriented, twisted fibrils that measure 8–12 mm in diameter
Fibrils accumulate first in the mesangium and later in the subendothelial space, producing long silver-positive bundles that extend perpendicularly to the basement membrane beneath podocytes (spicules) (Fig. 34.22)
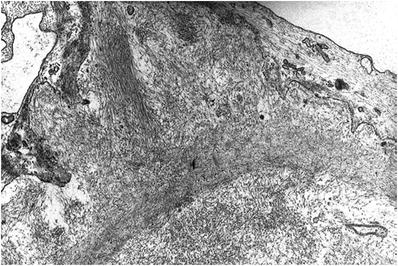
Fig. 34.22.
Electron micrograph of capillary wall amyloid with subepithelial “spicules”.
Light Chain Deposition Disease
♦
A systemic disease caused by overproduction of a monoclonal immunoglobulin light chain (usually κ), which deposits in various organs
Microscopic
♦
Light microscopy
The glomeruli contain eosinophilic mesangial nodules similar to the nodules seen in diabetic nephropathy and in amyloidosis
Like amyloid, the mesangial nodules in light chain deposition disease are nonargyrophilic and stain weakly with PAS (Fig. 34.23)
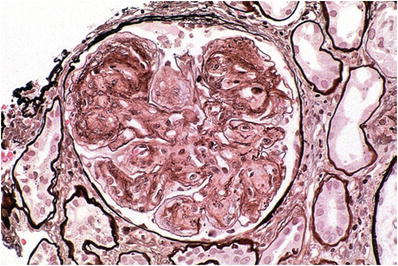
Fig. 34.23.
Light chain deposition disease with pale-staining mesangial nodules on silver stain.
♦
Immunofluorescence microscopy
There is bright linear staining of tubular basement membranes for κ light chains and less intense staining of the glomeruli; other immunoglobulins may be present rarely (Fig. 34.24)
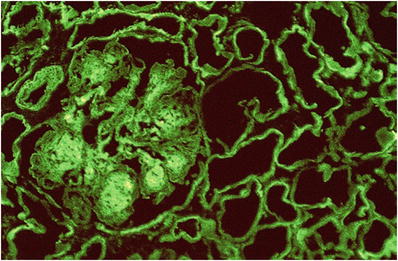
Fig. 34.24.
Bright tubular basement membrane and glomerular staining for kappa light chain in light chain deposition disease.
♦
Electron microscopy
Punctate granular deposits in the lamina rara interna of the glomerular basement membrane with extension into the lamina densa and lamina rara externa are less common
•
Mesangial deposits are less prominent
The deposits are more consistently present in the tubular basement membranes and have a similar granular appearance
Light chain proximal tubulopathy
♦
A paraprotein-related disease characterized by tubular dysfunction (Fanconi syndrome) as the result of resorbed free light chains
Microscopic
♦
Light microscopy
Features of acute tubular injury (see later) are often present, with tubular dilation and epithelial flattening. Protein resorption droplets may be present in some cases
♦
Immunofluorescence microscopy
Restricted granular kappa or λ staining is identified within tubular epithelial cells
♦
Electron microscopy
In cases with kappa light chains, epithelial cells contain pleomorphic rhomboidal to circular crystals (Fig. 34.25). These may be absent in instances with λ light chain restriction
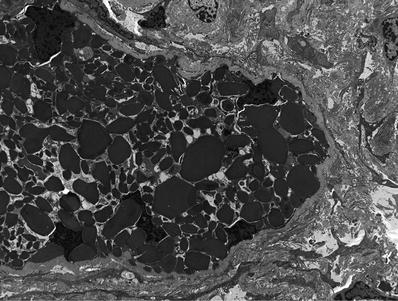
Fig. 34.25.
Rhomboidal crystals within tubular epithelial cell cytoplasm ultrastructurally in light chain proximal tubulopathy.
Cryoglobulinemic Glomerulonephritis
♦
Cryoglobulins are circulating immunoglobulins that precipitate on cooling and resolubilize on warming
♦
Three types of cryoglobulinemia have been described:
In type I cryoglobulinemia, the immunoglobulin is a single monoclonal immunoglobulin usually without associated antibody activity
In type II cryoglobulinemia, a monoclonal immunoglobulin (usually IgM-κ) is directed against polyclonal immunoglobulin (usually IgG)
In type III cryoglobulinemia, a polyclonal immunoglobulin is directed against a polyclonal immunoglobulin
Clinical
♦
Type I cryoglobulinemia is most often associated with lymphoproliferative disorders such as multiple myeloma, Waldenstrom macroglobulinemia, and chronic lymphocytic leukemia
♦
Mixed cryoglobulinemia (types II and III) occurs in a variety of settings, including lymphoproliferative disorders, collagen-vascular diseases, and infections including hepatitis B and C and Epstein–Barr virus
♦
Several studies have demonstrated an increased incidence of hepatitis C virus infection in patients with mixed cryoglobulinemia
Antibody to hepatitis C virus has been detected in serum from 91% to 98% of patients with mixed cryoglobulinemia and hepatitis C virus RNA in 81%
Microscopic
♦
Light microscopy
The acute glomerular lesion of mixed cryoglobulinemia is a diffuse proliferative glomerulonephritis with prominent subendothelial and intraluminal PAS-positive, silver-negative deposits
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


