and Alena Skalova2
(1)
Departamento de Ciências Biomédicas e Medicina, Universidade do Algarve, Faro, Portugal
(2)
Department of Pathology, Medical Faculty Charles University, Plzen, Czech Republic
2.1 Calculi
Sialolithiasis is a common, possibly the most common, disease of salivary glands and may occur in any of the salivary glands and at almost any age, childhood included [8]. The vast majority though occur in major salivary glands, the submandibular gland being the site in 80–90 % of cases. Less than 20 % are found in the parotid gland and only 1–2 % in the sublingual gland and minor salivary glands. The most common site in minor salivary glands is the anterior part of the mouth, especially close to the labial commissure of the upper lip. Although rare in minor salivary glands, more than 100 such cases have been described in the literature. Minor salivary gland calculi seldom exceed 5 mm. The long, tortuous, upward path of the submandibular duct and the thicker, mucoid secretions of the submandibular gland may be the reason for its greater tendency to form calculi. The size of salivary calculi can range from a few millimetres to so-called giant salivary gland calculi being larger than 15 mm. The giant calculi are rare and are most frequently encountered in the submandibular gland, and stones as large as 50 mm are reported [1–3, 13]. The aetiology of calculi is little known, and the exact mechanism of formation is unknown. Smoking appears to be unrelated to the formation of calculi [6]. Microcalculi are however present in serous acinar cells, striated duct cells, lumina and interstitium of normal parotid and submandibular glands. They may form in autophagosomes in parenchymal cells and pass into the lumina to be expelled in the saliva. Their size is approximately 25 μm when intracellular and somewhat larger when located in lumina. Microcalculi are hence found in normal glands and are unrelated to calculi. Microcalculi are related to the age of the patient whereas calculi appear to be secondary to sialadenitis and related to the duration of symptoms [5]. Calculi are predominantly composed of calcium phosphate (hydroxyapatite) with smaller amounts of ammonium, magnesium, potassium and iron. Deficit of crystallisation inhibitors such as myoinositol hexaphosphate (phytate) could be an important etiologic factor [7]. Contrary to what may be a common view, residents of hard water areas are not at increased risk of developing salivary calculi [14]. Multiple and also bilateral parotid sialoliths have been reported in patients with parotid MALT lymphoma and Sjögren’s syndrome [15, 17]. The first report of detection of ochronotic pigments in acinar cells and lumina in the submandibular gland, as well as the presence of a large calculus, was described in a patient with alkaptonuria (AKU). AKU is an autosomal recessive disease leading to widespread deposition of oxidised homogentisic acid polymer, primarily in the joints [16].
Recurrent episodes of swelling and pain associated with eating are the most typical sign, however less so in cases of parotid calculi compared to submandibular gland calculi. The symptoms gradually disappear a couple of hours after eating. The diagnosis can be made by history, by palpation and by means of sialography and zeroradiography. The management of calculi implies nowadays a minimally invasive approach consisting of sialoendoscopy, extracorporeal shock-wave lithotripsy, fluoroscopically guided basket retrieval or intraoral stone removal. Indication for a complete removal of the submandibular gland is becoming uncommon [4, 9, 11, 12]. The cutoff point for endoscopic removal is usually 5–6 mm in stone diameter [10].
A significant percentage of submandibular glands removed for sialolithiasis show normal histological findings apart from the calculus itself. However, the ducts containing calculi may show squamous metaplasia, and adjacent glandular parenchyma may show varying degrees of inflammation, atrophy and fibrosis. If present, the chronic inflammatory infiltrate tends to be periductal, and in the vicinity of a calculus, distended ducts may be seen. The lumina of the acini become visible from shrinkage and atrophy of the acinar cells, which lose their zymogen granules and may then resemble intercalated duct epithelium. Inasmuch as 30 % of all cases of chronic sclerosing sialadenitis of the submandibular gland (Küttner tumour), an association with calculus can be observed (see below; Sialadenitis). In certain circumstances, dystrophic calcification of the submandibular gland occurs with large areas of the gland replaced by calcium deposits. Whether this represents a progressive enlargement of a solitary calculus or multicentric calcification remains unclear. A distended duct may rupture, and mucous as well as fragments of the calculus may leak out into the glandular parenchyma, often provoking a foreign body reaction with giant cells (Fig. 2.1).
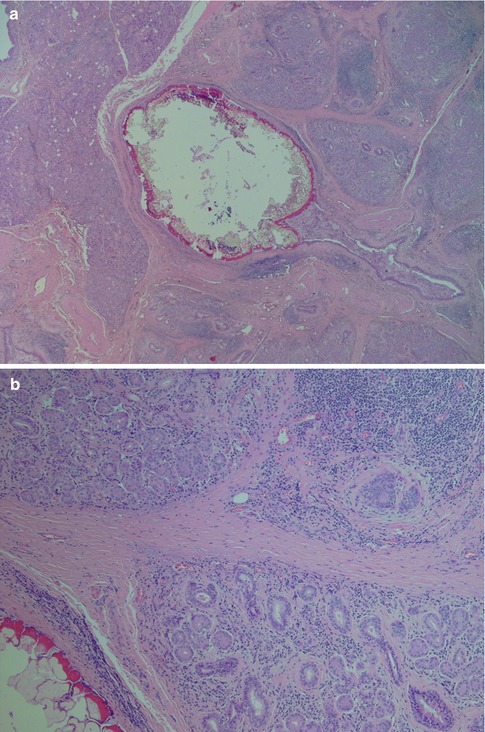
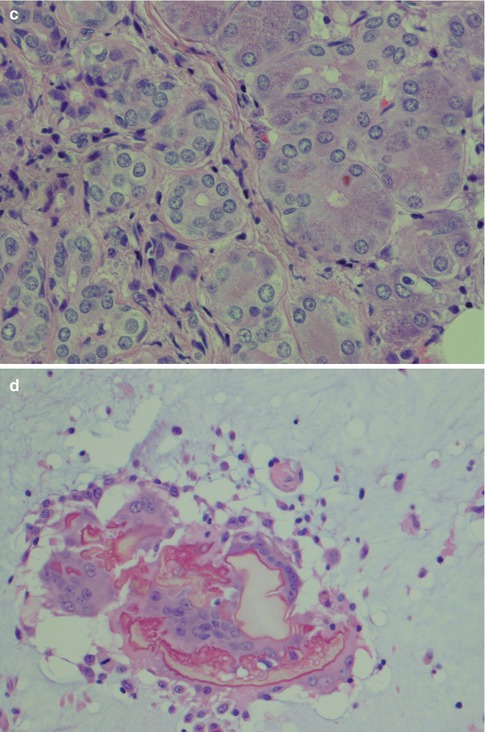
Fig. 2.1
Submandibular gland calculus. (a) A 2.5 mm large stone, partly fragmented during processing. (b) Atrophy of glandular parenchyma, inflammation and fibrosis in the vicinity of the calculus (lower left). (c) Normal acini with cytoplasmic granules (right) and atrophy of acinar cells (left). The atrophic acinar cells have lost their zymogen granules, partly have visible lumina and resemble intercalated ducts. (d) Fragments of a calculus with surrounding multinucleated giant cells floating in extraductal mucin
2.2 Cysts
Mucocele, mucous retention cyst, salivary duct cyst, ranula, lymphoepithelial cyst and branchial cyst are cystic lesions whose nomenclature and pathogenesis have been controversial in the past. Obstruction of salivary ducts by calculi or mucous plugs with consequent dilatation has previously been considered as the cause of most of these cysts. The term mucocele is applied to a cyst that lacks proper epithelial lining, whilst those that have epithelium are called mucous retention cysts, and a different aetiology appears likely. These non-neoplastic cysts form together approximately 6 % of all salivary gland lesions, and many may clinically resemble neoplasms. In a series of 1,303 salivary gland cysts, 75 % were located in minor salivary glands (85 % mucoceles and 15 % mucous retention cysts), 10 % were salivary duct cysts (retention cysts) of the parotid gland, 5 % were lymphoepithelial cysts (predominantly of the parotid gland) and the remaining 10 % were ranulas and other cystic lesions [50].
Mucoceles are lined by fibrous tissue, histiocytes and granulation tissue rather than epithelium and are also referred to as extravasation cysts. The vast majority (50–80 %) of mucoceles are found in the lower lip and, then in decreasing frequency in the floor of the mouth, cheek and palate. Less than 5 % occur in upper lip, and the possibility of a cystic salivary gland tumour should always be considered when examining cystic lesions from this site [26, 50]. Bhaskar and associates demonstrated experimentally in rats that duct ligation could not produce a lesion similar to the human mucous retention cyst lined with epithelium. By cutting the duct, extravasation of mucous was induced and the cyst formed did not have any epithelial lining [21]. Like the experimentally induced cysts, mucoceles lined by granulation tissue are considered to be caused by trauma rather than by obstruction. The walls are poorly defined and composed of granulation tissue (Fig. 2.2). Inside the cyst, there is mucin and inflammatory cells, predominantly histiocytes. Sometimes the cyst disappears and is replaced by a mass of histiocytes, known as Hamperl’s granulomas (Fig. 2.3) [31]. On rare occasions, a mucocele may become infected and develop into a pyocele. The high incidence of mucoceles in the lip and the rarity of cysts with epithelial lining in this location further support the theory of Bhaskar and associates. Furthermore, if calculi and obstruction leading to retention of mucous were the major cause for these cysts, one would expect a much higher incidence in the major salivary glands where calculi are so much more common. It has also been suggested that some of the intraoral mucoceles are instead caused by traumatic destruction of a large amount of glandular acini rather than a duct. The mucous from the disintegrated cells would form a pool, which in time is surrounded by a connective tissue capsule also containing remnants of parenchyma of the affected lobule. As this parenchyma degenerates, a cyst is eventually formed having the histological features of a mucocele. Cystic lip salivary gland neoplasms, e.g. acinic cell carcinoma and canalicular adenoma, may clinically mimic mucoceles and constitute an important differential diagnosis, particularly on the upper lip. Mucoceles may be multiple which not uncommonly so in cases of superficial mucoceles. A superficial mucocele is located immediately below the epithelium and may create a blister that can rupture. The salivary duct opens into the blister-like formation, and they are often found in the region of the palatoglossal fold [24, 39, 46, 47].
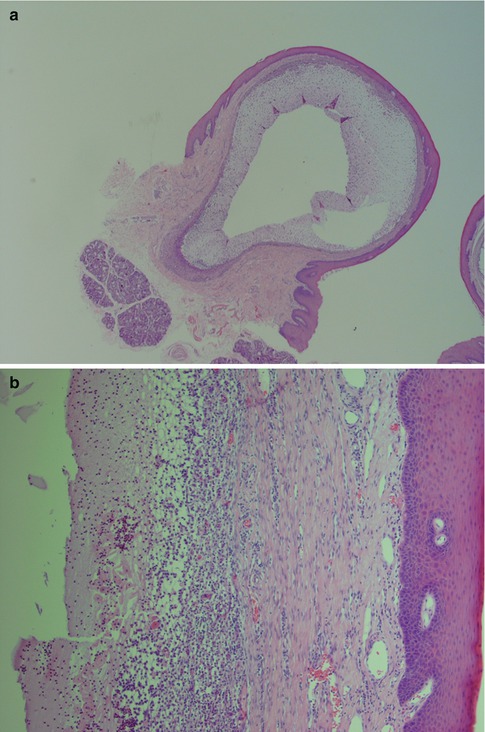
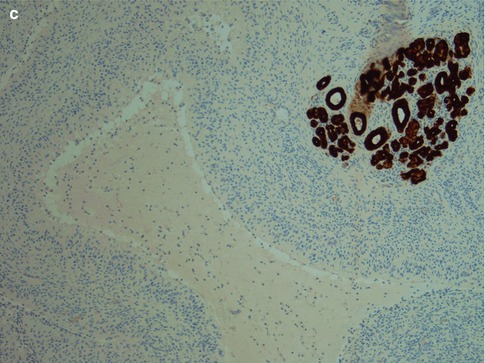
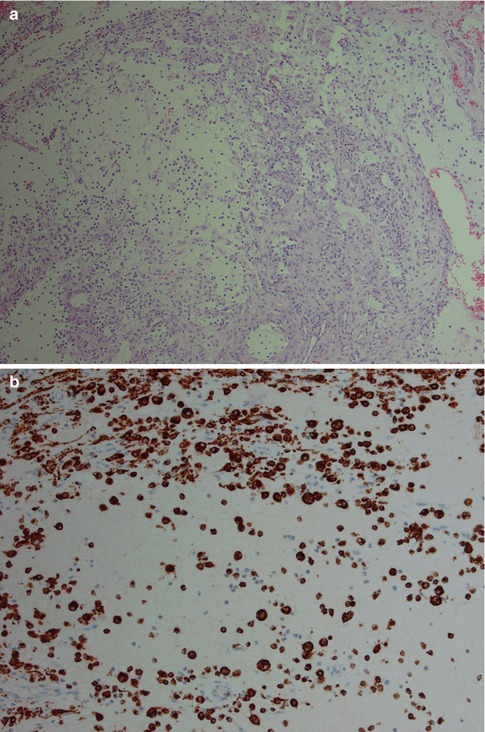
Fig. 2.2
(a) Lower lip mucocele. (b) The wall of the cyst is devoid of epithelial lining and consists of fibrous tissue and inflammatory cells. The lumen is partly filled with mucous and histiocytes. (c) Immunostain with pancytokeratin highlights the lack of epithelial lining; mucocele centre
Fig. 2.3
(a) Development of Hamperl’s granulomas consisting of accumulation of histiocytes and granulation tissue. (b) Immunostain for CD68 shows numerous histiocytic cells in Hamperl’s granuloma
Mucous retention cysts are lined with an epithelium that is in continuity with the lining of the duct and can hence be regarded as a dilatation of a duct. They are not caused by trauma but by obstruction, and in contrast to mucoceles, they are more common in major salivary glands. The epithelium is often of a flattened columnar type, or metaplastic squamous type. Occasionally the epithelium consists of columnar cells with oncocytic change (Fig. 2.4). The epithelium typically stains positive for CK8/18, CK5/6 and CK7 but negative for CK20 and TTF-1, similar to the normal salivary ductal epithelium. Occasionally, there can be focal positivity for TFF-1 as is the case with branchial cysts (Fig. 2.5) (see below; Branchial cysts). The ‘false’ positivity for TTF-1 may cause a dilemma in the differential diagnosis of thyroglossal duct cyst, should the lesion be situated in the midline, e.g. in the tongue, and no thyroid follicles are present in the wall. The glandular tissue surrounding a mucous retention cyst may show acinar atrophy, visible lumina and ductal dilatation. Sometimes the retention cyst may be large, filled with clear fluid and easily ‘shelled out’ from the parotid tissue. These cysts are thin-walled 05–1.5 mm and consist of fibrous tissue, more or less nor remnants of salivary tissue and lined with flattened cuboidal cells (Fig. 2.6). Salivary duct cyst (sialocyst) is another name for a mucous retention cyst of the parotid that also occasionally occurs in the submandibular gland. In contrast to mucoceles, only about 20 % of mucous retention cysts are localised in the lower lip, and they show an age peak in the seventh and eighth decade. Mucoceles, on the other hand, are most common in the second, third and fourth decade. Ranula is a cyst of the floor of the mouth, and the swelling resembles the vocal or air sacs of the frog (rana means frog and ula little). It appears as a blue, dome-shaped, fluctuant swelling in the floor of the mouth, usually lateral to the midline. The histogenesis may be dysgenetic disturbances of the differentiation of the sublingual gland duct system, or obstruction, or trauma causing extravasation of mucous from the duct into the glandular tissue. Therefore some ranulas may have epithelial lining as in retention cysts, whilst other lack epithelium as in mucoceles. Clinically two varieties of ranula have been defined. The simple ranula is a conventional mucous retention cyst that lie in the submucosal layers of the floor of the mouth. The so-called plunging ranula extends beyond the mucous membranes into the floor of the mouth and into the fascial planes of the neck. The plunging ranula is almost always a mucocele without epithelial lining and is likely the result of mucous extravasation. The most common origin of ranulas is the deeper areas of the body of the sublingual gland and its main duct (Bartholin’s ducts), followed by a lesser degree from the ducts of Rivini (minor sublingual ducts), and less frequently from the opening of Wharton’s duct (submandibular duct) [20].
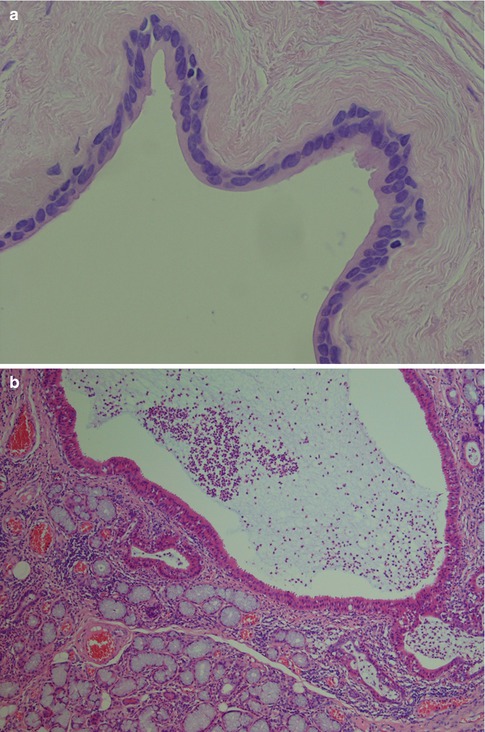
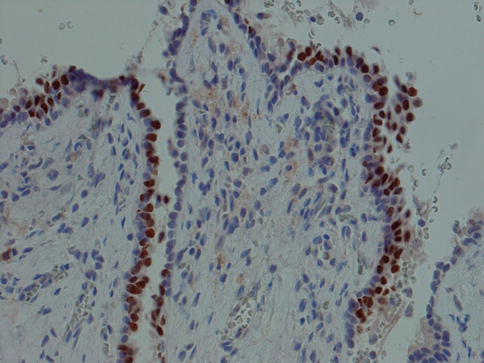
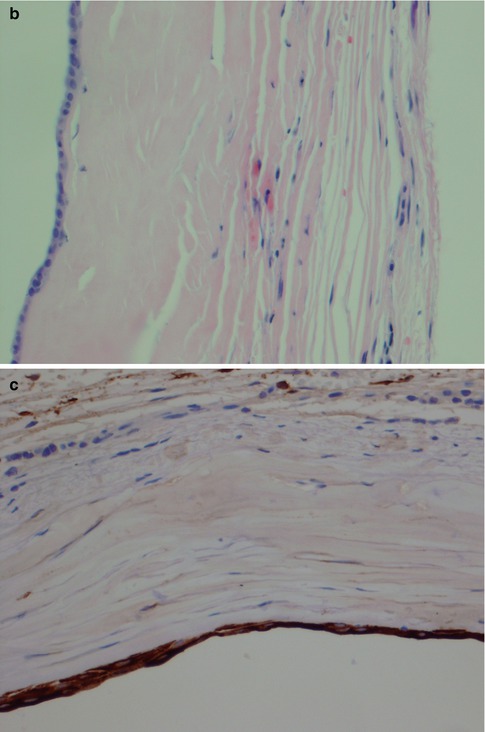
Fig. 2.4
(a) Mucous retention cyst where the epithelial lining consists of flattened cuboidal cells. (b) Another mucous retention cysts where the epithelium consists of 1–2 layers of cuboidal/columnar cells with oncocytic change
Fig. 2.5
A mucous retention cyst where many cells of the epithelium are positive for TTF-1
Fig. 2.6
(a) A large thin-walled retention cyst. (b) Epithelial lining of the same thin-walled cyst. (c) The same thin attenuated epithelial lining is CK7 positive
Lymphoepithelial cysts are caused by obstruction of salivary ducts by diffuse lymphoid infiltrate in the glandular tissue. The cysts arise from the intralobular duct system rather than from intraparotid lymph nodes and present as either as nodular or diffuse enlargements. Oral lymphoepithelial cysts may occur but are rare and are located almost exclusively in the floor of the mouth although a few cases have been reported arising in the tongue. The cysts are lined with squamous cell epithelium, which particularly in oral cases shows parakeratosis. Although most often stratified squamous epithelium, there may be smaller areas where the lining consists of cuboidal cells or respiratory tract type epithelium. Surrounding the cyst lining there are accumulations of lymphocytes often arranged in a follicular pattern. The parotid lymphoepithelial cysts have also been repeatedly reported in HIV-infected patients, and it has been estimated that multiple and bilateral parotid lymphoepithelial cysts are encountered in 3–6 % of HIV-infected patients. HIV-associated salivary gland disease as such is defined as the presence of xerostomia and/or swelling of the salivary glands. Lymphoepithelial cysts are regarded as an early manifestation of HIV infection, and HIV testing is therefore recommended for patients with benign lymphoepithelial cysts. One autopsy study of a series of 100 AIDS patients revealed that six patients had lymphoepithelial cysts [27, 32, 33, 37, 40, 49, 54, 55]. Although the parotid glands are by far the most common site, the submandibular gland can be affected. A rising prevalence of ranula which mirrors that of Kaposi’s sarcoma has recently been described in Zimbabwe. In a study of 83 patients with ranula, 88.5 % tested HIV positive with 95 % of them in the 0–10-years of age. A sublingual ranula may therefore be considered another HIV/AIDS associated lesion in Zimbabwe, particularly in children [23]. There is a continuous histological spectrum of changes within the salivary parenchyma, starting with lymphoid stroma infiltrate (nowadays thus many cases associated/induced by HIV) and evolving to characteristic lymphoepithelial duct lesions with a basal cell proliferation and to fully developed cysts. There is no participation of myoepithelial cells. The lymphatic infiltration of the salivary parenchyma shows an explosive hyperplasia with prominent follicular reticular dendritic cell network and numerous intrafollicular CD8+ lymphocytes. The intrafollicular reticular dendritic cell has shown to strongly express HIV-1 major-core protein and HIV-1 virus, indicating that most reticular dendritic cells actively replicate the HIV-1 virus [37]. A clinicopathological analysis of 64 cases where no viral infection was confirmed showed that the lymphoid stroma had neither the usual follicular distribution of T/B cells nor lymph sinus structures [56]. Apparent regression of lymphoepithelial cysts following antiviral treatment has been reported [41], and also alcohol injection sclerotherapy is proven to be an effective and cheap procedure for patients who do not qualify for antiretroviral therapy [44]. Salivary gland fistulae are almost always caused by trauma and most commonly affect the parotid gland. The discharge of saliva occurs into the skin and very rarely into the oral mucosa.
Branchial cysts are abnormalities in the development of the branchial apparatus and may occur in the vicinity of the parotid or submandibular glands. Cysts developing from the first branchial cleft appear in the preauricular region. The majority of branchial cysts arise from the second branchial cleft or the cervical sinus or branchial pouch. They are found deep to the sternomastoid muscle or along its anterior border, most commonly at the level of the angle of the mandible [18]. The amount of surrounding lymphoid tissue and type of epithelium tend to vary depending on the age of the patient and how long-standing the cyst has been. In a study of 97 lateral neck cysts arisen from the developmental remnants of the second branchial cleft, cysts in young patients (under 4 years) and of recent symptomatic presentation in older patients were lined by respiratory tract epithelium with scant surrounding lymphoid tissue. In adolescents, the respiratory epithelial lining alternates with multilayered pseudostratified epithelium containing lymphocytes and Langerhans cells. The lymphoid tissue is more prominent and with hyperplastic secondary follicles. In adults and longer-standing symptomatic cysts, the epithelium is mostly squamous in type with interspersed areas of respiratory epithelium. The respiratory epithelium is essentially CK18 positive and CK14 negative, whilst the multilayered and squamous epithelium becomes essentially CK14 positive and CK18 negative. These changes occur in branchial cysts with developing age, and the stratification and epidermoid change and lymphoid hyperplasia are thought to be due to postnatal immunological stimulation [48]. The epithelial lining of a branchial cyst has recently been demonstrated to be immunopositive for thyroid transcription factor 1 (TTF-1), in spite of no developmental relationship to the thyroid, lung or brain [35]. The reported single case is the exception confirming the rule that branchial cysts are TTF-1 negative. However, occasionally also the epithelium of mucous retention cysts stain positively for TTF-1 (see above, Fig. 2.5). The branchial cysts differ from lymphoepithelial cysts in several aspects, e.g. the epithelium in the latter is proper stratified squamous epithelium rather than a mixture of squamous epithelium and respiratory epithelium, or metaplastic squamous epithelium, as seen in branchial cysts. The branchial cyst epithelium bears resemblance to the tonsillar crypt epithelium (Fig. 2.7). There is also a salivary gland tissue in the vicinity of the lymphoepithelial cyst which is usually not the case in the branchial cyst. Most branchial cysts derive from the second branchial cleft and therefore located at the level of the angle of the mandible. Furthermore, the branchial cyst is usually circumscribed by a fibrous pseudocapsule in between which and the epithelial lining there is a variable amount of lymphoid tissue. There are germinal follicles as well as lymphoid aggregates, the latter often as small nodules protruding into the lumen similar to what is seen in Warthin tumour (Fig. 2.8). The epithelium is often proliferative, and numerous nests of epithelium can be seen in the lymphoid tissue, features that are not commonly encountered in lymphoepithelial cysts. The proliferating epithelial nests are located in the lymphoid stroma and not infiltrating the surrounding fibrous tissue and should not be mistaken for carcinoma. A metastatic, cystic carcinoma will show areas of necrosis which is not seen in the proliferating epithelial nests of a branchial cyst. The epithelium and nests can show a very high proliferative index as measured by Ki-67, and there may be reactive atypia present but few mitotic figures. The Ki-67 positivity in branchial cyst epithelium is restricted to the basal half of the epithelium, and the epithelium also shows maturation. The epithelium shows positivity for pancytokeratin markers, CK5/6 and CK8/18 but is negative for CK7 and CK20. The lymphocytic infiltrate consists of an approximate equal amount of B and T lymphocytes, and in the latter population CD4+ cells predominate. The hyperplasia with prominent follicular reticular dendritic cell network and numerous intrafollicular CD8+ T lymphocytes characteristically seen in lymphoepithelial cysts are not present in the branchial cyst. A fair number of CD8+ T cells are present in branchial cysts but the majority are CD4+ T cells. The T-cell CD4:CD8 ratio has been estimated to be 2:1 [48] (Fig. 2.9).
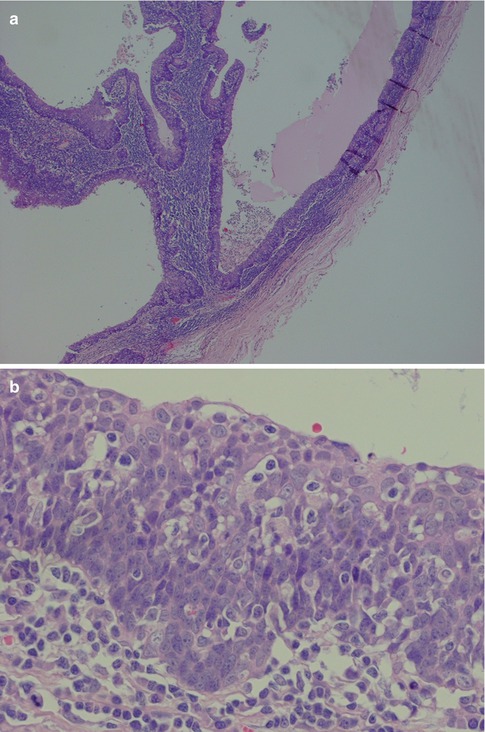
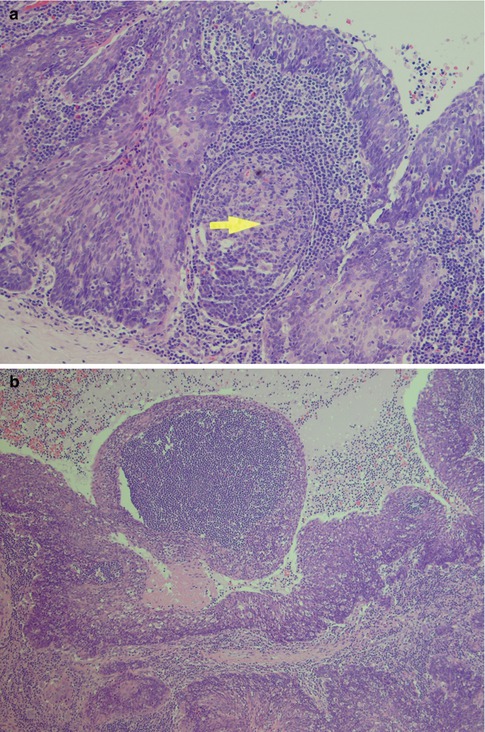
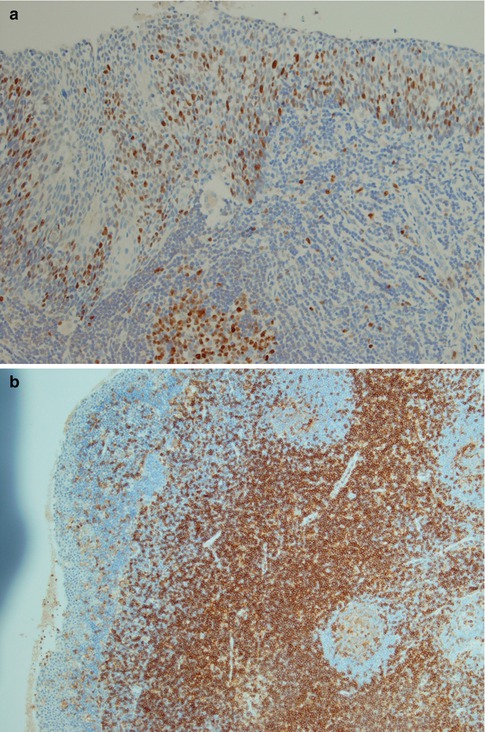
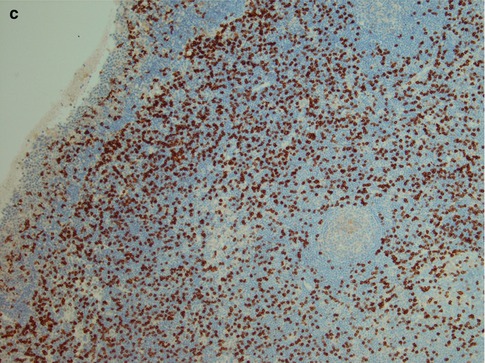
Fig. 2.7
Branchial cyst in a 59-year-old man. (a) Low magnification showing the surrounding fibrous tissue (right) and lymphoid tissue beneath the epithelium. (b) Metaplastic squamous epithelium with a very ‘crowded’ (tonsillar crypt) appearance
Fig. 2.8
(a) Cyst wall with fibrous pseudo-capsule (bottom) and epithelial lining (top). In between the proliferating and budding epithelium, there is a germinal centre (arrow). (b) Nodule protruding into the lumen and filled with a lymphoid aggregate. Note epithelial islands in the lymphoid stroma (bottom)
Fig. 2.9
(a) High Ki-67 index primarily in the lower half of the epithelium. Germinal centre present (bottom centre). (b) The majority of T cells are CD4 positive. (c) There are many CD8 positive cells, however, considerably fewer than CD4 positive cells
The development of malignancy in salivary cysts is a very rare event. Examination of 1,661 salivary gland cysts revealed only one case of malignancy, a mucoepidermoid carcinoma in a parotid duct cyst [51]. The existence of primary branchiogenic carcinoma may still be controversial, but the literature does provide some few but well-documented cases. New criteria for the diagnosis of primary branchiogenic carcinoma were proposed by Khafif and associates in 1989 and are currently the most recognised in the literature [22, 30, 34, 38]. The strict criteria for the establishment of a diagnosis of primary branchiogenic carcinoma should include [34]:
1.
Location of the tumour in the anatomical region of the branchial cleft cyst or sinus as previously defined [42]
2.
Histological appearance of the tumour consistent with its origin from branchial vestiges, i.e. squamous cell carcinoma
3.
Presence of the carcinoma within the lining of an identifiable epithelial cyst
4.
Identification of transition from the normal squamous epithelium of the cyst to carcinoma
5.
Absence of any identifiable primary malignant tumour after exhaustive evaluation of the patient
Khafif et al. reviewed all 67 cases published in the English literature at that time, and 41 of the 67 cases were definitely ruled out as carcinomas of branchial cysts. Although only eight of the remaining 26 cases satisfied the original Martin’s criterion of 5-year follow-up without evidence of primary carcinoma elsewhere, 14 patients had incontrovertible evidence showing a cyst with epithelial dysplasia progressing to squamous cell carcinoma [34].
Polycystic (dysgenetic) disease is an exceedingly rare developmental disorder so far reported primarily in females and characterised by painless enlargement of one or more often both parotid glands. So far only approximately 15 cases have been described in the literature. One case affecting both submandibular glands and one case of accessory salivary gland involvement have been described, whilst the remaining cases were parotid lesions almost exclusively in females [29, 36, 43]. Polycystic disease is not associated with any clinical abnormality of salivation or with any apparent anomaly of the other salivary glands. The cystic changes are due to the developmental malformation of the intercalated duct system. The lobular structure is preserved, but much of the parenchyma is replaced by a honeycomb of cysts of varying sizes. The cysts are lined by a single layer of flattened or cuboidal cells and contain secretion products, spheroliths and microliths. Residual parenchyma, interlobular septa and excretory ducts lie between the cysts. Dysgenic polycystic disease resembles cystic anomalies of other organs, e.g. the lungs and pancreas [19, 25, 28, 45, 52, 53].
2.3 Sialadenitis and Sialadenosis
Sialadenitis is caused by viruses, bacteria, immune disorders and also by radiotherapy. It may be acute or chronic, and on rare occasions, granulomatous as in, for example, cases of tuberculosis, but even Crohn’s disease with salivary gland involvement has been described [99]. Any inflammatory process may cause swelling of the salivary gland and thereby mimic a neoplasm. The presence of inflammatory cells distinguishes sialadenitis from sialadenosis, the latter being a non-inflammatory lesion associated with endocrine disorders, chronic alcoholism, liver cirrhosis, malnutrition, etc.
Several viruses, e.g. Cocksackie, cytomegalovirus, influenza A, and Echo virus, may cause a parotid swelling, but mumps is the most common. Mumps is a systemic disease and biopsy specimens from a salivary gland with mumps are rare as the diagnosis usually is a clear-cut clinical one with serologic tests. It is the most common viral sialadenitis and primarily affects the parotid glands but also to a certain degree the submandibular glands. The virus may be isolated from the urine a week after the salivary gland manifestations. Deafness may appear inasmuch as 4 % of adult cases of epidemic mumps parotitis; however, although severe it is usually unilateral and reversible. The acute vertigo affecting young children with mumps is often overlooked [80]. Bacterial sialadenitis is caused by a retrograde contamination of the salivary ducts and parenchyma by bacteria from the oral cavity. Stasis of salivary flow through the ducts promotes acute suppurative infection, and systemic dehydration is a well-known cause of salivary stasis. Chronic bacterial infections like tuberculosis or syphilis may involve the entire oral mucosa but rarely the minor salivary glands and are most often confined to the parotid glands. Acute bacterial infections, on the other hand, usually affect the entire oral cavity including the minor salivary glands. Many surgical procedures require that patients postoperatively are routinely without oral intake and consequently then being without regular salivary stimulation. Acute bacterial sialadenitis occurs in as many as 1 in 1,000 postoperative patients [60]. This acute suppurative infection is so closely linked to prior surgery that it is referred to as nosocomial (or postoperative) parotitis, and causative factors appear to be severe primary salivary gland hyposecretion related to old age, dehydration, oral inactivity and drugs. A mortality as high as 27 % has been reported [90]. However, other studies performed 10–15 years later reported no deaths, an improvement likely due to advances in antimicrobial therapy and fluid management. Nosocomial acute bacterial sialadenitis may be a better term than nosocomial parotitis as it affects not only the parotid gland but the submandibular gland in 30 % of cases. Nosocomial sialadenitis is known to occur 1–15 weeks following surgery, but most commonly appears within 2 weeks after the surgical procedure, with a peak incidence between the fifth and seventh postoperative day [93]. As for other acute forms of sialadenitis, Staphylococcus aureus and Streptococcus viridans are the most common bacteria [97]. Juvenile recurrent parotitis is a rare salivary gland inflammatory disease that most frequently involves children before puberty with a peak incidence between the ages of 3 and 6. The parotid swelling is sudden in onset, often tender and sometimes accompanied by a fever. Juvenile recurrent parotitis is usually unilateral but may occasionally be bilateral. Cystic dilatations of the interlobular ducts have been observed and the aetiology remains unknown [91].
The histology of the salivary gland specimen excised in clinical sialadenitis will show a non-specific chronic inflammation, and the aetiology will in most cases remain unknown; however, many cases are associated with sialolithiasis. Chronic sialadenitis does not have the histological features of lymphoepithelial sialadenitis (see below Sect. 2.4, Lymphoepithelial Sialadenitis. LESA), and the composition and distribution of the inflammatory infiltrate suggest that infectious agents may have triggered the inflammatory response. The histology is characterised by varying degrees of periductal and diffuse infiltrate of lymphocytes and plasma cells, with or without acinar atrophy, and sometimes also fibrosis. CD4+ T cells are the predominant lymphocytes [105]. The accumulation of lymphocytes like a rim around a salivary gland tumour is known as tumour-associated lymphoid proliferation (TALP). Whether this lymphocytic accumulation is a defensive response to tumour growth or constitutes some kind of lymphoepithelial background remains unclear [61, 81]. The fibrosis can be pronounced such as in cases with chronic sclerosing sialadenitis of the submandibular gland (see below). The fibrosis may also surround the remaining ducts concentrically like onionskins, particularly seen in parotid tissue adjacent to a neoplasm (Fig. 2.10). The regeneration of salivary gland tissue during and after inflammatory conditions has previously been thought to be due to proliferation of a hypothetical population of uncommitted ductal stem cells. It has been shown, however, that the proliferative indices were significantly increased in chronic sialadenitis in mature acinar, intercalated ductal cells and myoepithelial cells without evidence of proliferation of an additional cell population. This profound capacity for intrinsic glandular regeneration from differentiated cells represents a new way of regarding regeneration and may offer new therapies for improvement of impaired salivary flow. In spite of the intrinsic cellular proliferation, chronic sialadenitis eventually leads to various degrees of glandular atrophy with loss of acinar cells but preservation of intercalated ducts and myoepithelial cells [82].
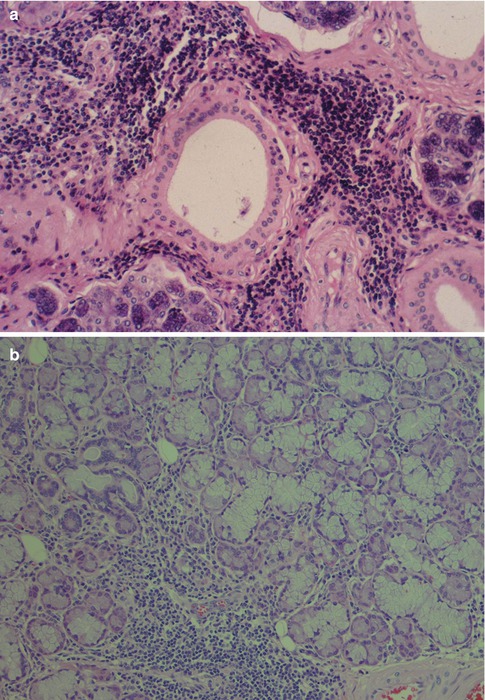
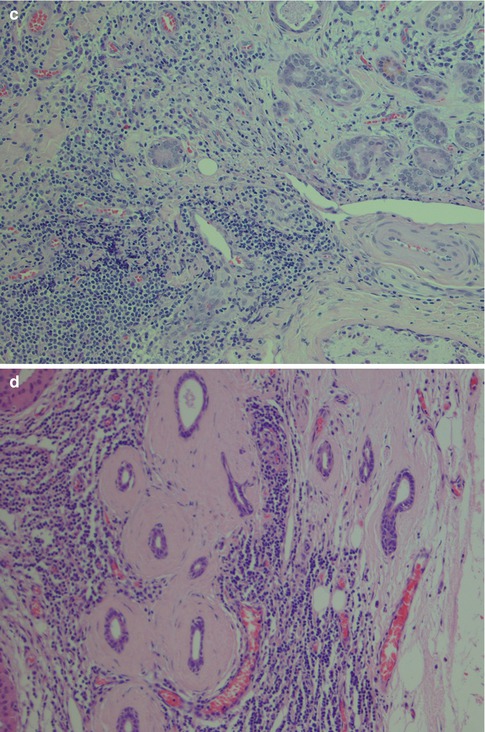
Fig. 2.10
(a) Periductal sialadenitis. (b) Sialadenitis with a mild to moderate chronic inflammatory infiltrate but no acinar atrophy or fibrosis. (b) More pronounced inflammation with replacement and atrophy of acinar cells that have lost most of their zymogene granules; mild fibrosis. (c) Sialadenitis with acinar atrophy and onionskin-like fibrosis
Granulomatous sialadenitis is recognised in tuberculosis, in sarcoidosis and, very occasionally, in other in infections and lesions such as cat scratch disease, Wegener’s granulomatosis and Churg-Strauss allergic granulomatosis. This type of granulomatous sialadenitis is rare, whilst granulomas caused by extravasation of mucin are not an uncommon incidental finding. A study of 469 major salivary glands excised for various reasons 57 (12 %) were found to contain granulomas, most frequently in the submandibular gland in association with calculous duct obstruction. These granulomas consist of foamy macrophages admixed with neutrophils and lymphocytes and foreign body giant cells but may also have epithelioid cells. The granulomas contain extracellular mucin on PAS staining [108, 111]. The histological appearance is very similar to Hamperl’s granuloma, a reaction caused by extravasation of mucin in cases of minor salivary gland mucocele [78, 101]. A very rare variant of granulomatous sialadenitis, xanthogranulomatous sialadenitis, has been described. The histology is similar to that of granulomatous sialadenitis except for the absence of extracellular mucin, nor are there any Michaelis-Gutmann bodies present as in malakoplakia. Most often it represents reactive changes secondary to another lesion, especially metaplastic Warthin tumour, but primary xanthogranulomatous sialadenitis without any other identifiable lesion exists [58, 96]. In 2013, Schaller and associates reported a case of granulomatous necrotising submandibular sialadenitis as a limited variant of Wegener granulomatosis [98].
Chronic sclerosing sialadenitis is a specific inflammatory process in the submandibular gland resulting in a firm and swollen gland that clinically may not be distinguished from a neoplasm. In 1896, Küttner [88] reported four cases of submandibular fibrosis and inflammation. Küttner observed an association with sialolithiasis and suggested that the sialadenitis arose from oral inflammation and the calculi subsequently developed from an irregularity of the ductal lining, in a lobule obstructed by inflammation or in a bacterial deposit. Due to the validity of Küttner’s report and the tumour-like clinical appearance of the lesion, the eponymous Küttner tumour is used as a synonym for chronic sclerosing sialadenitis of the submandibular gland. Four degrees of severity of inflammation can be distinguished [100]. In Stage 1, there is focal sialadenitis with lymphocytic inflammation primarily around ducts but also diffuse parenchymal inflammation. In Stage 2, there is more marked diffuse inflammation and periductal fibrosis. During Stage 3, there is a reduction of the glandular parenchyma, ductal proliferation and development of lymph follicles, some with reactive germinal centres. In Stage 4, the lobular architecture is partly destroyed and resembles liver cirrhosis (Fig. 2.11). In Stage 4, one may also see lymphoepithelial lesions (epimyoepithelial islands) and the question arises whether chronic sclerosing sialadenitis can be defined as sialadenitis with transition into autoimmune sialadenitis. There are reports claiming that helper/inducer cells significantly dominate over suppressor/cytotoxic T cells, and hence showing a similar pattern to antigenically stimulated lymph nodes [63, 84]. Although immune-mediated processes are very likely in its pathogenesis, the exact immunopathological processes of chronic sclerosing sialadenitis remain unknown. Contrary to previous studies, Tiemann and associates have demonstrated that cytotoxic T cells, and not T helper subsets, clearly predominate, and especially so in the periductal and periacinar infiltrates. They further showed that in about a third of cases, T-cell receptor gamma chain rearrangements showed a monoclonal pattern, oligoclonal pattern in another third of cases, and polyclonal in the remaining third. These findings indicate that this type of sialadenitis may will be the result of an immune process triggered by intraductal agents [109].
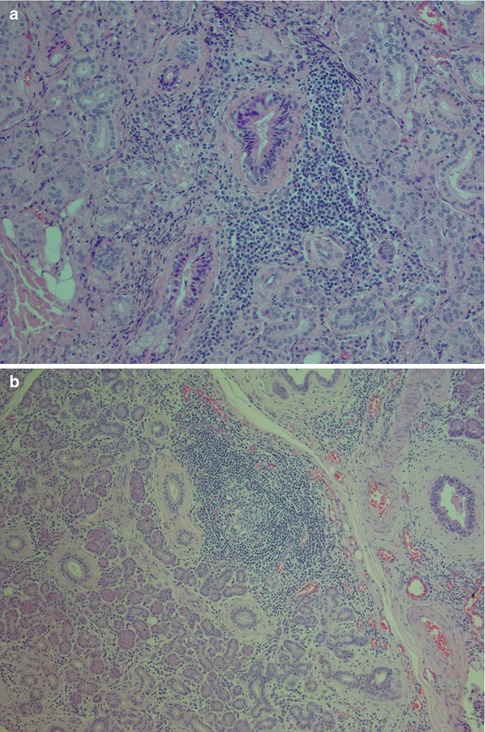
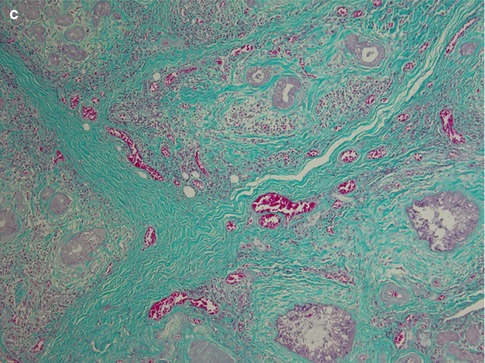
Fig. 2.11
Chronic sclerosing sialadenitis of the submandibular gland (Küttner tumour). (a) Stage 1 with periductal inflammation but preserved glandular parenchyma. (b) Submandibular gland with marked lymphoid infiltrate with formation of reactive germinal centre. (c) Marked sclerosing fibrosis and reduction of glandular parenchyma (Trichrome)
It has been suggested that Küttner tumours can be characterised by their abundant IgG4-positive plasma cells. Immunohistochemistry showed the proportion of IgG4/IgG-positive plasma cells to be more than 45 % in chronic sclerosing sialadenitis but less than 5 % in controls. This abundance of IgG4-positive plasma cells may therefore be a useful diagnostic tool in distinguishing Küttner tumour from other forms of sialadenitis [86]. Chronic sclerosing sialadenitis of both submandibular glands as well as both parotid glands has been described in a patient with IgG4-related sclerosing pancreatitis [95]. There is hence increasing support for regarding chronic sclerosing sialadenitis as part of the spectrum of IgG4-related systemic disease. IgG4-related disease is a fibroinflammatory condition unrecognised in medical science until the last decade. IgG4-related sclerosing disease is characterised by focal or multifocal involvement by tumefactive plasma cell-rich inflammation associated with fibrosclerosis and also elevated serum titer of IgG4. IgG4-related sclerosing disease shows a good response to steroid therapy. The prototypic lesion of IgG4-related disease is lymphoplasmacytic sclerosing (autoimmune) pancreatitis. IgG4-related disease has been described in almost all organs, and the head and neck is emerging as one of the most moccon sites after the pancreatobiliary system [59, 62, 66–68, 71, 75]. Out of the 12 cases with submandibular chronic sclerosing sialadenitis described by Kitagawa and associates [86], five had associated sclerosing lesions in extrasalivary glandular tissue and referred to as systemic type. The extrasalivary sites involved consisted of (1) sclerosing cholangitis, (2) sclerosing pancreatitis and (3) in one case lacrimal gland swelling. Patients with the systemic type also frequently had eosinophilia and elevations of γ-globulin and IgG in serum. The remaining seven patients only had involvement of the submandibular gland(s) and referred to as localised type. Chronic sclerosing sialadenitis is hence characterised by atrophy or destruction of glandular lobules, marked lymphoplasmacytic infiltration admixed with irregular and dense fibroplasias and obliterative phlebitis. More than 45 % of infiltrating IgG-positive plasma cells are of IgG4-type which is in sharp contrast to sialadenitis seen in cases of sialolithiasis and Sjögren’s syndrome where less than 5 % of IgG-positive plasma cells are of IgG4-type [86]. Although labial salivary gland biopsy is minimally invasive and widely used for the diagnosis of Sjögren’s syndrome, a submandibular gland biopsy is recommended for accurate diagnosis of IgG4-related disease. In a study of 15 patients with IgG4-related disease, histology of submandibular biopsy revealed fibrosis in all cases but present in only one of the salivary gland biopsies [104].
Subacute necrotising sialadenitis is a rare non-specific inflammatory, self-limiting lesion of intraoral salivary glands. It usually presents as a painful, non-ulcerated swelling of the palate of short duration, some days to a month only. The aetiology is unknown but infections and allergic responses could be possibly the causes. Subacute necrotising sialadenitis has certain features in common with necrotising sialometaplasia (see below, Sect. 2.5, Necrotising Sialometaplasia), but it affects a considerably younger age group, also predominantly men, than does necrotising sialometaplasia. Most patients are in the second or third decades of life. The histology also differs to that of necrotising sialometaplasia with no lobular necrosis, nor any squamous metaplasia. Further there is a rather marked, diffuse mixed inflammatory infiltrate often with eosinophils. Although the two lesions may be part of a spectrum of inflammatory changes, most authors do recognise subacute necrotising sialadenitis as a separate entity [74, 76, 110, 112].
Irradiation–induced sialadenitis is a frequent condition in the head and neck cancer patients and affects primarily the submandibular gland, but also the parotid and minor salivary glands may be affected depending on the irradiation field applied. The glands are usually received from neck dissections after primary surgery and radiotherapy. Irradiation-induced sialadenitis is histologically characterised by gradual fibrosis and atrophy of glandular parenchyma and a varying degree of accompanying inflammatory infiltrate. Amongst the lymphocytes and besides the atrophic acini, there may be scattered macrophages and monocytic cells, as well as occasional Langerhans giant cells and epithelioid cells. The severity of the morphological changes varies. The longer time interval between the end of radiotherapy and the time of gland resection, the more pronounced changes are. Early it can be only a mild periacinar lymphocytic infiltration and periductal fibrosis with focal acinar atrophy. With progression there will be a pronounced diffuse lymphocytic infiltration, still predominantly in the periacinar area, an accentuated periductal and parenchymal fibrosis, and a moderate acinar atrophy. The most severe changes show only a slight focal lymphocytic infiltration, but the destruction of lobular architecture marked parenchymal loss and also sclerosis (Fig. 2.12). The majority of the lymphocytes consists of CD3+ T cells, whilst CD20+ B cells are mainly restricted in small nodules. CD8+ T cells predominate over CD4+ T cells [106]. There is hence a rather significant dominance of cytotoxic T cells contrary to chronic non-irradiation sialadenitis where CD4+ subsets predominate, and which are mainly periductal and not periacinar. The serous cells are the most sensitive and the first to be destroyed which has been linked to their content of secretory granules. These granules are rich in heavy metals. Due to the metal-catalysed induction of lipid peroxidation by ionising radiation, it may be possible that the membranes enclosing these granules show enhanced sensitivity to radiation damage by forming redox systems [57]. Although acinar cells are well-differentiated non-dividing cells, radiation causes interphase cell death in rhesus monkey parotid acinar cells within hours of receiving a radiation dose as low as 2.5 Gy [103]. Recent studies, however, have shown that there is no cell loss during the first days after irradiation, whilst saliva flow is dramatically diminished and hence the theories of massive apoptosis and leakage of granules are questioned. These studies indicate the mechanism of action behind the early effects is selective radiation damage to the plasma membrane of the secretory cells, disturbing muscarine receptor stimulated watery secretion. Later damage would mainly be explained by mitotic cell death of progenitor cells leading to a hampered replacement capacity of secretory cells [87].
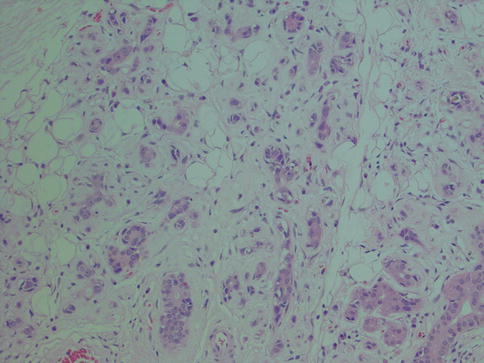
Fig. 2.12
Irradiation sialadenitis with pronounced atrophy and replacement of glandular parenchyma by fibrous connective tissue and fat. Relatively few inflammatory cells present
Sialadenosis is a non-inflammatory lesion associated with metabolic and secretory disorders of the salivary glandular parenchyma. It is clinically characterised by a recurrent painless bilateral swelling, usually of the parotid glands. Its aetiology is unknown, but sialadenosis is often associated with systemic diseases like endocrine disorders, particularly diabetes mellitus, but also ovarian and thyroid insufficiencies, and with liver cirrhosis, chronic alcoholism and malnutrition. Sialadenosis associated with anorexia nervosa and bulimia is well recognised, and involvement of minor salivary glands has also been reported [69, 77, 94]. Sialadenosis of minor salivary glands as an isolated event is very rare, if it at all exists. Primary neuropathy of the autonomic nervous system has been suggested as a possible causative factor, seen as a demyelinating polyneuropathy, and leading to disordered secretion on the acinar cells due to lengthening of the storage phase of the secretory granules and an arrest of protein synthesis [72, 85]. Autonomic neuropathy may cause functional myoepithelial insufficiency. The observed loss and thinning of the myofilament component in the myoepithelial cells would result in a reduction of the mechanical support for the acini and thereby play a role in the pathogenesis of sialadenosis [83]. It has also been proposed that antidiuretic hormone (ADH) and the water channels, the aquaporins (AQP), could be implied in the pathomechanism of sialadenosis. The first water channel was identified in 1988 as a membrane protein from erythrocytes and renal tubules and was named aquaporin-1 [70]. Since then at least ten more aquaporins have been identified and AQP2 (from renal collecting ducts) is known to be regulated by ADH. The application of ADH leads to upregulation of AQP2 leading to renal water retention [64]. AQP5 is of particular interest with regard to salivary gland disorders being a water channel protein that is selectively expressed in salivary, respiratory and lacrimal tissues. The AQP5 is localised on apical surface of the acinar salivary cells and in patients with sialadenosis and Sjögren’s syndrome, the AQP5 protein is upregulated and abnormally distributed towards the acinar cellular base, respectively [65, 89, 92, 102]. Upregulation of AQP5 by frequent ADH application, as described in a patient with central diabetes insipidus, and consequent altered expression levels of AQP5, may therefore explain a disturbance of the cell volume regulation in sialadenosis. Immunohistochemical studies by Teeymoortash and associates have demonstrated stronger AQP5 signals at the apical plasma membrane and also that the distribution at the apical region differed compared to normal salivary gland tissue [107]. Upregulation of AQP5 and possible development of sialoadenosis could also be a reaction to a chronic reduction in free water due to frequent diuresis (diabetes mellitus) and diuresis due to alcohol-dependent inhibition of ADH secretion (alcohol abuse) [79, 92]. The histological hallmarks of sialadenosis are hypertrophy of acinar cells and absence of inflammatory infiltrate. The serous acinar cells become enlarged with the nuclei are displaced towards the basal part of the cell. The cytoplasm shows either granular pattern or a vacuolar transformation. The swollen cells often cause a mild compression of the acinar duct system (Fig. 2.13). The normal average size of parotid acinar cells are 30–40 μm, whilst in sialadenosis the diameters are enlarged to 50–70 μm, in some cases to as much as 100 μm [73].
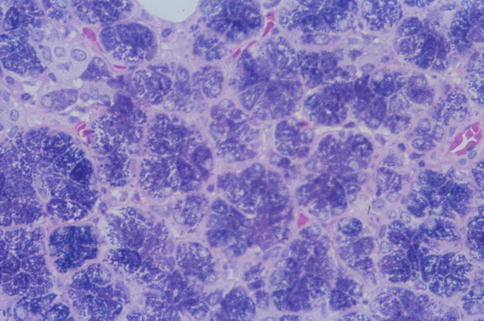
Fig. 2.13
Sialadenosis with enlarged granular acinar cells. Compressed duct is seen upper left
2.4 Lymphoepithelial Sialadenitis
Reactive lymphoid proliferations of the salivary glands include cystic lymphoid hyperplasia in HIV-positive patients and lymphoepithelial sialadenitis, LESA. In 1892, Mikulicz described a case of a patient with unusual bilateral painless swelling of the lacrimal glands and all of the salivary glands [141]. The histological examination of involved glands showed features of what today is recognised as lymphoepithelial sialadenitis. After Mikulicz’s report, clinicians began to describe bilateral salivary gland swellings as Mikulicz syndrome; however, histology showed many of these cases to represent salivary involvement of a number of other diseases, e.g. tuberculosis, malignant lymphoma, sarcoidosis, etc. Mikulicz syndrome is thus not a specific pathological entity but a hotchpotch of entities. In the early 1950s, Godwin reported a series of ten patients with salivary lesions consisting of lymphoid hyperplasia and epithelial alterations. The term benign lymphoepithelial lesion, BLEL, was introduced to replace salivary gland lesions previously known as Mikulicz syndrome [127]. Soon after Godwin’s report Morgan and Castleman gave a detailed description of the characteristically altered salivary ducts and named them epimyoepithelial islands [143]. With the introduction of immunohistochemistry in the 1970s and 1980s and the recognition that some of the lymphoid proliferations were in fact low-grade lymphomas, and not benign, the term BLEL has fallen out of favour. The term myoepithelial sialadenitis, MESA, was introduced in the 1980s for the histopathological lesion [149]. Later, however, it has been demonstrated that the non-lymphoid cells are not myoepithelial cells but basal and ductal epithelial cells, and the term lymphoepithelial sialadenitis, LESA, is more appropriate. Likewise, the so-called epimyoepithelial island is likely better called lymphoepithelial lesions [130, 133, 144]. Lymphoepithelial sialadenitis may also be associated with other salivary lesions, e.g. sialadenitis, adenomas and lymphomas, but is closely associated with Sjögren’s syndrome. Hence, patients with lymphoepithelial sialadenitis may or may not have Sjögren’s syndrome, but, on the other hand, patients with Sjögren’s syndrome almost invariably have, or will develop, salivary gland lesions with histological features of lymphoepithelial sialadenitis.
Lymphoepithelial sialadenitis is histologically defined by heavy lymphocytic infiltrate associated with destruction of salivary acini but persistence of ductal epithelium. The lymphoid proliferation involves infiltration of ductal epithelium by lymphocytes of marginal zone or monocytoid B-cell type, forming the so-called epimyoepithelial islands. Despite the widely accepted term ‘epimyoepithelial islands’, the often postulated participation of myoepithelial cells in duct lesions in benign lymphoepithelial lesion cannot any longer be supported. Myoepithelial cells (basket cells) of acini and intercalated ducts are apparently not participating, and the ‘epimyoepithelial’ islands, i.e. lymphoepithelial islands, stem from hyperplasia of basal cells of striated ducts as evidenced, e.g. by immunopositivity for Ks8.12 and negativity for α-actin. The basal cells show aberrant differentiation into a multilayered and reticulated epithelium, characterised by a profound alteration of the cytokeratin pattern [133]. The earliest changes are periductal and perivascular infiltration of relatively small lymphocytes. They are accompanied by larger lymphocytes and plasma cells, and eventually the lymphocytic infiltrate becomes extensive and germinal centres may develop. There is progressive swelling and replacement of acini. Proliferation of striated ductal cells leads to formation of the so-called epimyoepithelial or lymphoepithelial islands in the lymphocytic infiltrate. Fibrosis and acinar atrophy may develop. The lymphocytic infiltrate contains B cells and including plasma cells that secrete antibody locally, but also numerous T cells, predominantly CD4+ helper cells. There is both preferential influx/homing of memory B cells and local proliferation that contribute to the pattern of lymphoepithelial sialadenitis in Sjögren’s syndrome [129]. There is a fully benign cellular infiltrate with or without an associated lymphoid follicular structure. There is no immunoglobulin light chain restriction in B cells. The distinction between LESA and extranodal marginal zone B-cell lymphoma of MALT type (EMZBCL) may be extremely difficult. Most EMZBCL arise in a setting of LESA in association with Sjögren’s syndrome, and the distinction between LESA/Sjögren’s syndrome and EMZBCL may on occasions not be possible without immunohistochemistry. Both contain lymphoepithelial islands and these islands are often invaded by B cells, a feature that is more prominent when invaded by neoplastic B cells in EMZBCL. In EMZBCL, monocytoid and centrocyte-like B cells tend to surround the lymphoepithelial islands forming ‘halos’, a feature not present in LESA. These surrounding B cells may coalesce into broad interconnecting sheets. Ductal dilatations are present in both lesions but usually more prominent in LESA. The lymphocytic infiltrate tends to be more diffuse and widespread in EMZBCL, whilst it in LESA often may retain a focal, almost nodular pattern. This nodular pattern is not to be confused with follicular lymphoma, in which the germinal centres will be positive not only for bcl-2 but for CD10 and bcl-6. Both LESA and EMZBCL contain benign germinal centres negative for bcl-2, but in EMZBCL the follicles may be colonised by neoplastic B cells expressing bcl-2. Light chain restriction supports the monoclonality of the B-cell lymphoma [116, 117, 132] (Fig. 2.14). The fully developed histological features of lymphoepithelial sialadenitis are usually seen in major salivary glands, whilst the histological features of Sjögren’s syndrome in minor salivary glands, often used for diagnostic purposes, are different (see below).
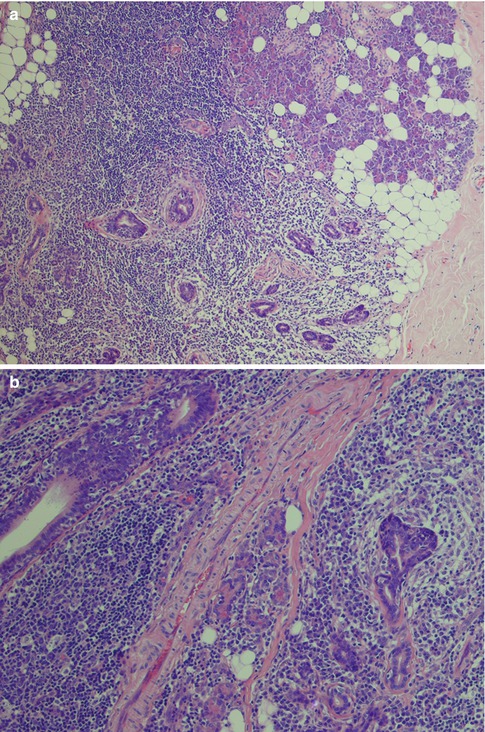
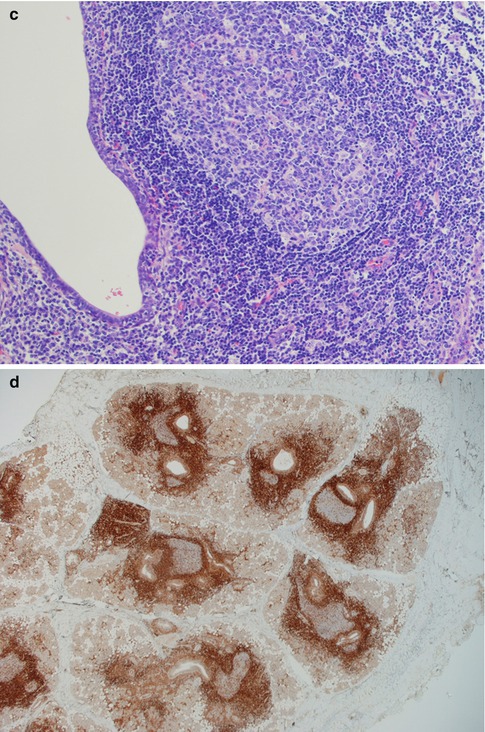
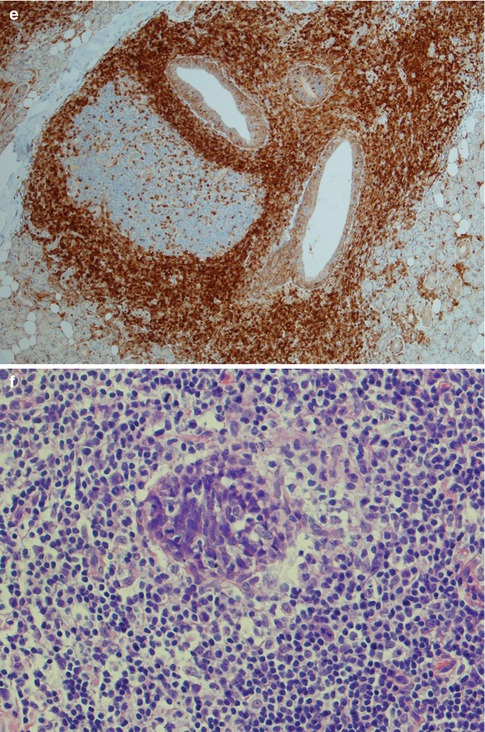
Fig. 2.14
Parotid lymphoepithelial sialadenitis in Sjögren’s syndrome (LESA). (a) Atrophic acini are replaced by a lymphocytic infiltrate. Preserved ducts (lower right) and early lymphoepithelial islands (centre). (b) Lymphoepithelial islands and a few preserved ducts (lower right). (c) Reactive germinal centre and dilated duct. (d) Bcl-2 immunostaining showing the lobular nature of inflammatory infiltrate within the salivary tissue, and germinal centres that are bcl-2 negative. (e) Higher magnification of bcl-2 negative germinal centre and dilated ducts partly infiltrated by B cells. (f) Lymphoepithelial island without the monocytoid B-cell halo typically seen in EMZBCL
In 1933, the Swedish ophthalmologist Henrik Sjögren described clinical and histological findings in 19 women, 13 of whom had probable rheumatoid arthritis, with dry mouth and eyes [151]. Sjögren’s syndrome (SS) is nowadays recognised as a complex autoimmune disorder characterised by mononuclear cell infiltration and decreased secretion in the salivary and lacrimal glands. Both cellular and humoral factors, such as autoreactive immune cells and autoantibodies, respectively, contribute to the expression of the disease. The population prevalence of Sjögren’s syndrome depends on what clinical classification criteria have been used, but there is a female preponderance (female to male ratio 9 to 1). The prevalence for Sjögren’s syndrome reaches from 0.5 % of all adult females when employing the Californian Fox Criteria up to 1–3 % of total population, when using the European Community criteria or the American European Consensus criteria [123]. It is one of the three most common autoimmune disorders but has hitherto received far less research and clinical attention than RA, SLE and systemic sclerosis [125]. Patients with SS have a 16–44-fold increased risk for the development of salivary gland or extrasalivary malignant lymphoma, and in approximately 5 % of patients, lymphoma will develop. Approximately 80 % are marginal zone/MALT type [123, 130, 154]. A small proportion of patients with Sjögren’s syndrome (<1 %) may have coexisting sarcoidosis [126]. The clinical signs of Sjögren’s syndrome, i.e. keratoconjunctivitis sicca and xerostomia, are results from immunologically mediated destruction of the salivary and lacrimal glands. Although SS formally consists of keratoconjunctivitis sicca, rhinitis sicca, pharyngolaryngitis sicca, parotid enlargement, xerostomia and polyarthritis, it implies in practice keratoconjunctivitis sicca, xerostomia and (rheumatoid) arthritis. Rheumatoid arthritis is the most common amongst the associated connective diseases, but SS can be associated with almost any autoimmune disease, and some patients have scleroderma, SLE, thyroiditis, polymyositis, vasculitis, etc. When there is no associated autoimmune disease, it is known as primary Sjögren’s syndrome, or sicca syndrome. Classification criteria for SS have been developed over the years, and in 2002, a consensus report with a revised version was published [155]. These classification criteria, the revised American European Consensus Group, have represented the golden standard for the last decade. Of these six criteria, two substantive and four objective, criterion IV, focal lymphocytic sialadenitis, and criterion V, anti-Ro/La antibodies, were considered to be the most specific disease markers for Sjögren’s syndrome. In 2012, the American College of Rheumatology endorsed a new set of preliminary criteria proposed by the Sjögren’s International Collaborative Clinical Alliance [150]. This new classification criteria substantially differ from the American European Consensus Group criteria, and a wish to reach a consensus agreement on universally accepted classification criteria for SS was voiced in 2013 [156].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


