Chapter 8 Nitrogen Metabolism
I. Biosynthesis of Nonessential Amino Acids
A. Overview
1. Eleven of the 20 amino acids are synthesized in the body (nonessential amino acids), and the remaining nine amino acids are required in the diet (essential amino acids).
2. Many of the nonessential amino acids are synthesized by transamination reactions, in which an amino group is added to an α-ketoacid to produce an amino acid.
a. Ten of the nonessential amino acids are derived from glucose through intermediates derived from glycolysis and the citric acid cycle.
3. Tyrosine is an exception in that it is derived from phenylalanine, which is an essential amino acid.
II. Removal and Disposal of Amino Acid Nitrogen
A. Overview
1. Removal of the α-amino group from amino acids is the initial step in the catabolism of amino acids.
5. Ammonia that is not converted to urea is carried to the kidneys for secretion into the urine (i.e., acidifies urine).
6. Hyperammonemia is associated with encephalopathy producing feeding difficulties, vomiting, ataxia, lethargy, irritability, poor intellectual development, and coma.
TABLE 8-1 Synthesis of Nonessential Amino Acids
| Amino Acid | Source of Carbon Skeleton | Comments |
|---|---|---|
| Alanine | Pyruvate | Transamination of precursor |
| Arginine | Ornithine | Reversal of arginase reaction in urea cycle |
| Asparagine | Oxaloacetate | Amide group from glutamine |
| Aspartate | Oxaloacetate | Transamination of precursor |
| Cysteine* | Serine | Sulfur group from methionine |
| Glutamate | α-Ketoglutarate | Transamination of precursor |
| Glutamine | α-Ketoglutarate | Amide group from free NH4+ |
| Glycine | 3-Phosphoglycerate | From serine through transfer of methylene group to tetrahydrofolate (THF) |
| Proline | Glutamate | Cyclization of glutamate semialdehyde |
| Serine | 3-Phosphoglycerate | Oxidation to keto acid, transamination, hydrolysis of phosphate |
| Tyrosine* | Phenylalanine | Hydroxylation by phenylalanine hydroxylase (tetrahydrobiopterin cofactor) |
* Can be synthesized only if methionine and phenylalanine are available from the diet.
B. Transamination and oxidative deamination (Fig. 8-1)
1. Step 1
a. Transamination entails the transfer of the α-amino group of an α-amino acid to α-ketoglutarate, producing an α-keto acid from the amino acid and glutamate from α-ketoglutarate (see Fig. 8-1, left).
b. Aminotransferases (transaminases) catalyze reversible transamination reactions that occur in the synthesis and the degradation of amino acids.
c. The two most common aminotransferases transfer nitrogen from aspartate and alanine to α-ketoglutarate, providing α-ketoacids that are used as substrates for gluconeogenesis.
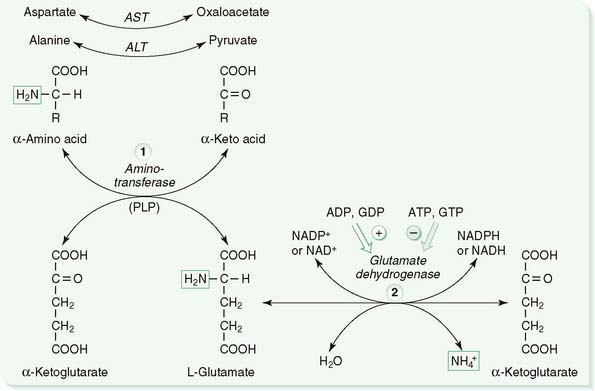
2. Step 2
a. Oxidative deamination of glutamate (the product of transamination) is the major mechanism for the release of amino acid nitrogen as charged ammonia (NH4+), and it occurs primarily in the liver and kidneys (see Fig. 8-1, right).
C. Urea cycle (Fig. 8-2)
1. The urea cycle functions mainly in the liver to convert highly toxic NH4+ to nontoxic urea.
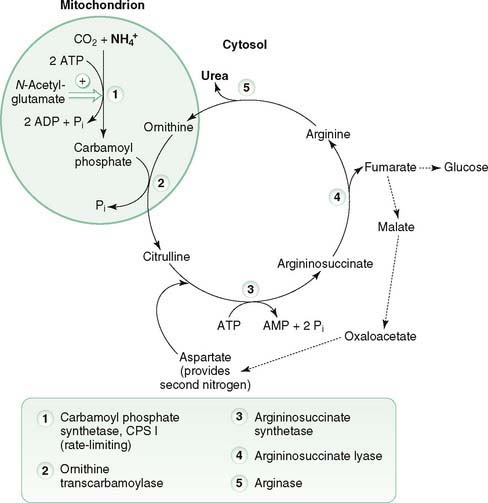
8-2 The urea cycle is located in the liver and is the primary mechanism for disposal of toxic ammonia.
2. Glutamate is the primary source of NH4+ that is used in the urea cycle; however, ammonia is produced from other sources that are metabolized by the cycle.
3. Urea cycle reactions occur in the mitochondrial matrix and cytosol.
a. Two mitochondrial reactions generate citrulline, which is transported to the cytosol.
(1) Step 1
(a) Carbamoyl phosphate synthetase I (CPS I) catalyzes the first, rate-limiting step in which NH4+ (contains the first nitrogen), CO2, and ATP react to produce carbamoyl phosphate (see Fig. 8-2).
b. Three cytosolic reactions incorporate nitrogen from aspartate to form ornithine, which reenters mitochondria, and urea, which leaves the cell.
D. Ammonia metabolism
1. Ammonia is primarily converted to urea, with the exception of ammonia derived from glutamine, which is used to acidify urine.
2. Sources of ammonia
a. Glutamate
(1) Ammonia is derived from oxidative deamination of glutamate by glutamate dehydrogenase (see Fig. 8-1).
d. Dietary protein
(1) Bacterial ureases release ammonia from amino acids in dietary protein and from urea diffusing into the gut.
(2) Depending on the pH, ammonia released by ureases is charged (NH4+) and nondiffusible through tissue or uncharged (NH3) and diffusible through tissue.
3. Ammonia produced in extrahepatic tissues is toxic and is transported in the circulation primarily as urea and glutamine.
5. Hyperammonemia results primarily from the inability to detoxify NH4+ in the urea cycle, leading to elevated blood levels of ammonia.
b. Acquired hyperammonemia most commonly occurs in alcoholic cirrhosis and Reye’s syndrome due to disruption of the urea cycle.
(1) In cirrhosis, the architecture of the liver is distorted, leading to shunting of portal blood into the hepatic vein or backup of blood in the portal vein (i.e., portal hypertension).
(2) Reye’s syndrome occurs primarily in children with influenza or chickenpox who are given salicylates.
c. Signs and symptoms of hyperammonemia include feeding difficulties, vomiting, ataxia, lethargy, irritability, poor intellectual development, and coma.
III. Catabolic Pathways of Amino Acids




A. Overview
1. Transamination of amino acid nitrogen produces carbon skeletons of amino acids as α-keto acids that enter intermediary metabolism at various points.
2. Amino acids are classified as glucogenic (degraded to pyruvate or intermediates in citric acid cycle), ketogenic (degraded to acetyl CoA or acetoacetyl CoA), or both glucogenic and ketogenic.
3. Carbon skeletons remaining after removal of the α-amino group from amino acids are degraded to intermediates that can be used to produce energy in the citric acid cycle or to synthesize glucose, amino acids, fatty acids, or ketone bodies.
4. Tyrosine is converted to catecholamines, thyroid hormones, melanin, and dopamine, and it is degraded to homogentisate.
5. Branched-chain amino acids—leucine, isoleucine, and valine—are degraded to branched-chain α-ketoacids that can enter the citric acid cycle.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
