Chapter 21 Neurology and the Neuromuscular System
Acetaminophen


MOA (Mechanism of Action) (Figure 21-1)
 Acetaminophen is neither a narcotic nor a nonsteroidal antiinflammatory drug (NSAID). It is in a drug class of its own called aniline analgesics.
Acetaminophen is neither a narcotic nor a nonsteroidal antiinflammatory drug (NSAID). It is in a drug class of its own called aniline analgesics.

 In contrast to NSAIDs and aspirin, acetaminophen is not an antiplatelet agent, nor does it possess antiinflammatory properties; therefore there are differences in the mechanism of action compared with aspirin or NSAIDs:
In contrast to NSAIDs and aspirin, acetaminophen is not an antiplatelet agent, nor does it possess antiinflammatory properties; therefore there are differences in the mechanism of action compared with aspirin or NSAIDs: Tissue selectivity: Acetaminophen demonstrates variable COX inhibition in different tissues. Of primary importance, it inhibits prostaglandin E2 (PGE2) production in the central nervous system (CNS), which is probably the primary mediator of its analgesic and antipyretic properties.
Tissue selectivity: Acetaminophen demonstrates variable COX inhibition in different tissues. Of primary importance, it inhibits prostaglandin E2 (PGE2) production in the central nervous system (CNS), which is probably the primary mediator of its analgesic and antipyretic properties.





Important Notes
 Therapeutic doses of acetaminophen have no effect on the cardiovascular and respiratory systems or platelet function and do not produce gastric irritation, erosion, or bleeding.
Therapeutic doses of acetaminophen have no effect on the cardiovascular and respiratory systems or platelet function and do not produce gastric irritation, erosion, or bleeding.Overdose
 Acetaminophen exposure is the most commonly reported drug exposure reported to U.S. poison control centers.
Acetaminophen exposure is the most commonly reported drug exposure reported to U.S. poison control centers.


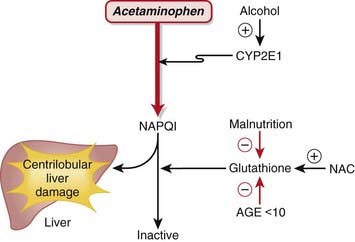
 The mechanism of injury is as follows:
The mechanism of injury is as follows: Acetaminophen is metabolized in the liver via CYP2E1 to N-acetyl-p-benzoquinone imine (NAPQI). NAPQI is highly reactive, electrophilic, and toxic.
Acetaminophen is metabolized in the liver via CYP2E1 to N-acetyl-p-benzoquinone imine (NAPQI). NAPQI is highly reactive, electrophilic, and toxic.

 NAC has been proposed to function as an antidote to acetaminophen via two additional mechanisms in addition to replenishment of GSH:
NAC has been proposed to function as an antidote to acetaminophen via two additional mechanisms in addition to replenishment of GSH: NAC can substitute directly for GSH because it has antioxidant properties and directly reduces NAPQI.
NAC can substitute directly for GSH because it has antioxidant properties and directly reduces NAPQI.



Evidence
Analgesia
Acetaminophen versus Placebo for Treatment of Osteoarthritis
 A Cochrane review in 2005 (seven studies) demonstrated that acetaminophen was superior to placebo in five of the seven randomized controlled trials (RCTs). A pooled analysis demonstrated a statistically significant but minimal difference that is of questionable clinical significance. The relative percent improvement in pain score from baseline was 5%, with an absolute change of 4 points on a 0-to-100 scale.
A Cochrane review in 2005 (seven studies) demonstrated that acetaminophen was superior to placebo in five of the seven randomized controlled trials (RCTs). A pooled analysis demonstrated a statistically significant but minimal difference that is of questionable clinical significance. The relative percent improvement in pain score from baseline was 5%, with an absolute change of 4 points on a 0-to-100 scale.Acetaminophen versus Nonsteroidal Antiinflammatory Drugs for Treatment of Osteoarthritis
 The same Cochrane review in 2005 (10 studies) demonstrated that acetaminophen was less effective overall than NSAIDs in terms of pain reduction, global assessments, and improvements in functional status. Patients taking traditional NSAIDS were more likely to experience an adverse GI event (relative risk [RR] 1.47). However, the median trial duration was only 6 weeks, which is too short to adequately assess adverse outcomes.
The same Cochrane review in 2005 (10 studies) demonstrated that acetaminophen was less effective overall than NSAIDs in terms of pain reduction, global assessments, and improvements in functional status. Patients taking traditional NSAIDS were more likely to experience an adverse GI event (relative risk [RR] 1.47). However, the median trial duration was only 6 weeks, which is too short to adequately assess adverse outcomes.Acetaminophen plus Codeine versus Placebo
 A Cochrane review in 2008 (26 studies, N = 2295 patients) of postoperative patients demonstrated significant differences for obtaining at least 50% pain relief over 4 to 6 hours, with a number needed to treat (NNT) of 2.2 for high doses (800 to 1000 mg acetaminophen plus 60 mg codeine) and smaller effect sizes for medium and smaller doses (as low as 325 mg acetaminophen with 30 mg codeine).
A Cochrane review in 2008 (26 studies, N = 2295 patients) of postoperative patients demonstrated significant differences for obtaining at least 50% pain relief over 4 to 6 hours, with a number needed to treat (NNT) of 2.2 for high doses (800 to 1000 mg acetaminophen plus 60 mg codeine) and smaller effect sizes for medium and smaller doses (as low as 325 mg acetaminophen with 30 mg codeine).Acetaminophen Plus Codeine versus Acetaminophen Alone
 A Cochrane review in 2008 (14 studies, N = 926 patients) of postoperative patients demonstrated that addition of codeine increased the proportion of participants achieving at least 50% pain relief over 4 to 6 hours by 10% to 15% and reduced the proportion of patients needing rescue medication by about 15%.
A Cochrane review in 2008 (14 studies, N = 926 patients) of postoperative patients demonstrated that addition of codeine increased the proportion of participants achieving at least 50% pain relief over 4 to 6 hours by 10% to 15% and reduced the proportion of patients needing rescue medication by about 15%.



Opioids






MOA (Mechanism of Action)
 The pain pathways in the body are very complex and only briefly summarized here. In short, opioids reduce the signaling and processing of pain pathways through a variety of receptor types, receptor locations, and complex interactions.
The pain pathways in the body are very complex and only briefly summarized here. In short, opioids reduce the signaling and processing of pain pathways through a variety of receptor types, receptor locations, and complex interactions.
 Exogenous opioids also bind multiple opioid receptors (Table 21-1). Opioid receptors are present in both the brain and the spinal cord, specifically:
Exogenous opioids also bind multiple opioid receptors (Table 21-1). Opioid receptors are present in both the brain and the spinal cord, specifically:




TABLE 21-1 Main Classes of Opioid Receptors
| Receptor | Action |
|---|---|
| Mu (µ) | |
| Kappa (κ) | |
| Delta (δ) |
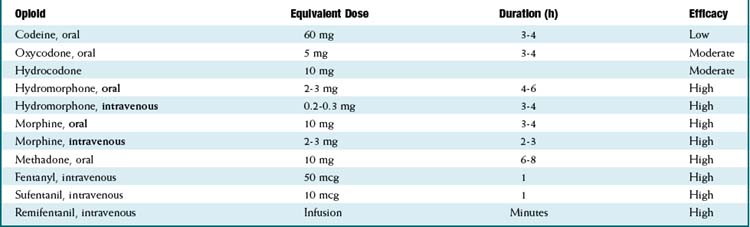
Pharmacokinetics (Table 21-2)
 For equivalency, note that:
For equivalency, note that:

 Important metabolites:
Important metabolites: Most opioids are metabolized by CYP3A4 and renally eliminated (except where the following information states otherwise).
Most opioids are metabolized by CYP3A4 and renally eliminated (except where the following information states otherwise). Morphine → morphine-3-glucuronide (90%), morphine-6-glucuronide (10%).
Morphine → morphine-3-glucuronide (90%), morphine-6-glucuronide (10%).• The glucuronides (being water soluble) are eliminated by the kidneys; renal disease can prolong and increase the effects of morphine.
• The glucuronides are water soluble; thus they do not readily cross the blood-brain barrier, but with high concentrations of drug, brain levels will increase.
• Morphine-6-glucuronide is more potent and longer acting than the parent compound morphine. Its accumulation can result in an increased and prolonged opioid effect







Side Effects
 Respiratory depression: All narcotics are powerful respiratory depressants. They can cause hypoventilation, hypoxia, and apnea. The effect is dose dependent. Low-efficacy opioids (codeine) are less likely to result in profound respiratory depression.
Respiratory depression: All narcotics are powerful respiratory depressants. They can cause hypoventilation, hypoxia, and apnea. The effect is dose dependent. Low-efficacy opioids (codeine) are less likely to result in profound respiratory depression.







Important Notes













Evidence
Opioids versus Nonsteroidal Antiinflammatory Drugs for Treatment of Renal Colic
 A systematic review in 2004 (20 trials, 1613 participants) found that both NSAIDs and opioids led to clinically important reductions in patient-reported pain scores. Pooled analysis of six trials showed a greater reduction in pain scores for patients treated with NSAIDs than with opioids. Patients treated with NSAIDs were significantly less likely to require rescue analgesia (RR 0.75). Most trials showed a higher incidence of adverse events in patients treated with opioids. Compared with patients treated with opioids, those treated with NSAIDs had significantly less vomiting (0.35).
A systematic review in 2004 (20 trials, 1613 participants) found that both NSAIDs and opioids led to clinically important reductions in patient-reported pain scores. Pooled analysis of six trials showed a greater reduction in pain scores for patients treated with NSAIDs than with opioids. Patients treated with NSAIDs were significantly less likely to require rescue analgesia (RR 0.75). Most trials showed a higher incidence of adverse events in patients treated with opioids. Compared with patients treated with opioids, those treated with NSAIDs had significantly less vomiting (0.35).Opioids for Treatment of Chronic Back Pain
 A systematic review in 2007 examined multiple questions pertaining to opioid treatment of chronic low back pain. 11 studies showed that there was significant variation in how opioids are prescribed for chronic low back pain. A meta-analysis of the four studies assessing the efficacy of opioids compared with placebo or a nonopioid control did not show reduced pain with opioids. A meta-analysis of the five studies directly comparing the efficacy of different opioids demonstrated a nonsignificant reduction in pain from baseline.
A systematic review in 2007 examined multiple questions pertaining to opioid treatment of chronic low back pain. 11 studies showed that there was significant variation in how opioids are prescribed for chronic low back pain. A meta-analysis of the four studies assessing the efficacy of opioids compared with placebo or a nonopioid control did not show reduced pain with opioids. A meta-analysis of the five studies directly comparing the efficacy of different opioids demonstrated a nonsignificant reduction in pain from baseline. With respect to risk of addiction, the prevalence of lifetime substance use disorders ranged from 36% to 56%; the prevalence of current substance use disorders was as high as 43%; and aberrant medication-taking behaviors (“drug seeking”) ranged from 5% to 24%. The authors found that the study was limited by retrieval and publication biases and poor study quality. No trial evaluating the efficacy of opioids was longer than 16 weeks.
With respect to risk of addiction, the prevalence of lifetime substance use disorders ranged from 36% to 56%; the prevalence of current substance use disorders was as high as 43%; and aberrant medication-taking behaviors (“drug seeking”) ranged from 5% to 24%. The authors found that the study was limited by retrieval and publication biases and poor study quality. No trial evaluating the efficacy of opioids was longer than 16 weeks.








α2 Agonists


MOA (Mechanism of Action)
 Glaucoma is characterized by increased intraocular pressure (IOP). Strategies to reduce intraocular pressure include reducing the production and secretion of aqueous humor and facilitating its drainage.
Glaucoma is characterized by increased intraocular pressure (IOP). Strategies to reduce intraocular pressure include reducing the production and secretion of aqueous humor and facilitating its drainage. The exact mechanism by which α2 agonists reduce IOP has not been established, but is likely multifactorial, employing several strategies:
The exact mechanism by which α2 agonists reduce IOP has not been established, but is likely multifactorial, employing several strategies:
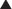

 The antihypertensive effect of α2 agonists is a result of inhibition of presynaptic release of vasoconstrictors such as norepinephrine. Recall that the α2 receptor is an autoreceptor (see Chapter 3). An autoreceptor is a receptor that when stimulated by an agonist, reduces release of transmitter into the synaptic cleft.
The antihypertensive effect of α2 agonists is a result of inhibition of presynaptic release of vasoconstrictors such as norepinephrine. Recall that the α2 receptor is an autoreceptor (see Chapter 3). An autoreceptor is a receptor that when stimulated by an agonist, reduces release of transmitter into the synaptic cleft.
Pharmacokinetics
 Brimonidine and apraclonidine are administered topically to the eye, and only a small amount reaches the systemic circulation.
Brimonidine and apraclonidine are administered topically to the eye, and only a small amount reaches the systemic circulation.




Contraindications
 Concomitant MAOIs: MAOIs prevent the breakdown of norepinephrine, and this could lead to elevated blood pressure.
Concomitant MAOIs: MAOIs prevent the breakdown of norepinephrine, and this could lead to elevated blood pressure.







Important Notes
 The ability of clonidine to modulate neurotransmitter release has also led to its increasing use in attention-deficit/hyperactivity disorder (ADHD).
The ability of clonidine to modulate neurotransmitter release has also led to its increasing use in attention-deficit/hyperactivity disorder (ADHD).


Evidence
In Primary Open Angle Glaucoma and Ocular Hypertension
 A 2007 Cochrane review of all medical interventions for glaucoma and ocular hypertension found three trials comparing brimonidine to timolol. There were no differences in visual field progression (glaucoma) or visual field defects (ocular hypertension) within 1 year. Timolol was better tolerated than brimonidine, as measured by the incidence of dropouts from drug-related adverse events (OR 0.21). There were no trials comparing brimonidine with placebo.
A 2007 Cochrane review of all medical interventions for glaucoma and ocular hypertension found three trials comparing brimonidine to timolol. There were no differences in visual field progression (glaucoma) or visual field defects (ocular hypertension) within 1 year. Timolol was better tolerated than brimonidine, as measured by the incidence of dropouts from drug-related adverse events (OR 0.21). There were no trials comparing brimonidine with placebo.
Inhaled Anesthetics


MOA (Mechanism of Action) (Figure 21-2)
 The primary site of action in causing CNS depression is most likely the γ-aminobutyric acid A (GABAA) receptor. Inhaled anesthetics activate these receptors.
The primary site of action in causing CNS depression is most likely the γ-aminobutyric acid A (GABAA) receptor. Inhaled anesthetics activate these receptors.
Pharmacokinetics
 As their name suggests, these drugs are administered only via inhalation. They are supplied in liquid form and then vaporized using very precise vaporizers that are part of the anesthetic machine, the anesthetic oxygen and air mixture is combined with calculated doses of the inhaled anesthetic.
As their name suggests, these drugs are administered only via inhalation. They are supplied in liquid form and then vaporized using very precise vaporizers that are part of the anesthetic machine, the anesthetic oxygen and air mixture is combined with calculated doses of the inhaled anesthetic.

 Inhaled anesthetics move through the body by dissolving in the blood and distributing into tissues; the important tissue interfaces include the following:
Inhaled anesthetics move through the body by dissolving in the blood and distributing into tissues; the important tissue interfaces include the following:
 Blood ∂ fat (and other vessel-poor tissues)
Blood ∂ fat (and other vessel-poor tissues)• When inhaled anesthetics are administered, the drug must enter the lungs, then the blood, then the brain.
• For inhaled anesthetics to be eliminated, drug must exit the brain and other tissues, be carried to the lungs, and then exhaled.
• Vessel-poor tissues are slow to take up and release drug. Therefore vessel-poor tissues can act as a sink and slowly absorb drug at the early parts of an anesthetic procedure (lowering the drug levels) but then at the end of a long anesthetic procedure can release drug, prolonging elimination of the drug.
 The solubility of an inhaled anesthetic affects the speed at which it is taken up by the body and exerts its action (speed of onset). The key point is that the partial pressure is the measure of the drug’s active form. If a drug has a high solubility, then a lot of drug needs to be absorbed into blood and tissues before the partial pressure starts to rise; this impedes the onset of action of the drug. If the solubility is low, then the partial pressure will rise quickly. Conversely, eliminating the drug follows the same rules, and high solubility correlates with slower elimination because more drug had to be dissolved into the body initially to achieve the desired partial pressure.
The solubility of an inhaled anesthetic affects the speed at which it is taken up by the body and exerts its action (speed of onset). The key point is that the partial pressure is the measure of the drug’s active form. If a drug has a high solubility, then a lot of drug needs to be absorbed into blood and tissues before the partial pressure starts to rise; this impedes the onset of action of the drug. If the solubility is low, then the partial pressure will rise quickly. Conversely, eliminating the drug follows the same rules, and high solubility correlates with slower elimination because more drug had to be dissolved into the body initially to achieve the desired partial pressure.• Solubility is described for the blood and is called the blood/gas coefficient. The smaller the number, the less soluble the drug is in blood and therefore the faster it can change its partial pressure (because only a small amount of drug actually needs to be dissolved into the blood). The agents are ranked from fastest to slowest (lowest to highest solubility coefficient) as follows:








Important Notes
 The potency of inhaled anesthetics is measured by the minimum anesthetic concentration (MAC), which is strictly defined as the dose of inhaled anesthetic required to prevent movement in 50% of the population in response to a surgical stimulus when no other drugs are administered. For example, the MAC of desflurane is 6%. Note that this definition is not the same as the dose required to keep someone asleep. Drugs such as opioids decrease the MAC requirements of a drug and are called MAC-sparing agents.
The potency of inhaled anesthetics is measured by the minimum anesthetic concentration (MAC), which is strictly defined as the dose of inhaled anesthetic required to prevent movement in 50% of the population in response to a surgical stimulus when no other drugs are administered. For example, the MAC of desflurane is 6%. Note that this definition is not the same as the dose required to keep someone asleep. Drugs such as opioids decrease the MAC requirements of a drug and are called MAC-sparing agents.



Advanced
 Nitrous oxide has a MAC value of 104%. Therefore, it is not potent enough to be a solo anesthetic agent, because delivering a mixture of 100% nitrous oxide would mean that no oxygen could be delivered and the patient would die of asphyxiation. The greatest concentration that is administered is about 60% to 70%—a MAC value of 0.6 or 0.7. For this reason, nitrous is used only as an adjuvant drug for general anesthesia and is added to one of the other volatile anesthetics.
Nitrous oxide has a MAC value of 104%. Therefore, it is not potent enough to be a solo anesthetic agent, because delivering a mixture of 100% nitrous oxide would mean that no oxygen could be delivered and the patient would die of asphyxiation. The greatest concentration that is administered is about 60% to 70%—a MAC value of 0.6 or 0.7. For this reason, nitrous is used only as an adjuvant drug for general anesthesia and is added to one of the other volatile anesthetics.




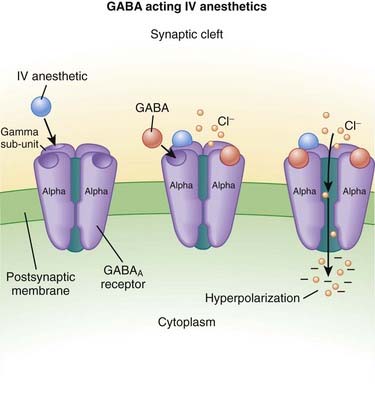
Intravenous Anesthetics



MOA (Mechanism of Action)
γ-Aminobutyric Acid (GABA) (Figure 21-3)
 GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC.
GABA is the major inhibitory neurotransmitter in the CNS. GABA binds to three different types of receptors: GABAA, GABAB, and GABAC. Binding of GABA to GABAA receptors leads to the opening of the chloride (Cl−) channel, facilitating Cl− influx and cellular hyperpolarization (making the inside more negative).
Binding of GABA to GABAA receptors leads to the opening of the chloride (Cl−) channel, facilitating Cl− influx and cellular hyperpolarization (making the inside more negative).




Pharmacokinetics
 Intravenous anesthetics work within one arm to brain circulation time; that is, if the drug is administered through an intravenous line in the arm, it is the time required to circulate back to the heart and then up to the brain (usually less than 1 minute).
Intravenous anesthetics work within one arm to brain circulation time; that is, if the drug is administered through an intravenous line in the arm, it is the time required to circulate back to the heart and then up to the brain (usually less than 1 minute).




Side Effects
 Respiratory depression: The respiratory center is depressed with GABA agonists. It is typical for a patient who is administered a dose large enough to induce unconsciousness to develop apnea (to completely stop breathing). For this reason, administration of intravenous anesthetics (regardless of dose) must always be performed in the presence of healthcare workers who are skilled in respiratory resuscitation and who have respiratory resuscitation equipment immediately available.
Respiratory depression: The respiratory center is depressed with GABA agonists. It is typical for a patient who is administered a dose large enough to induce unconsciousness to develop apnea (to completely stop breathing). For this reason, administration of intravenous anesthetics (regardless of dose) must always be performed in the presence of healthcare workers who are skilled in respiratory resuscitation and who have respiratory resuscitation equipment immediately available.



Important Notes

 Dosage: Some general concepts should be applied when choosing the dose of intravenous anesthetic. Administering a larger-than-required dose virtually guarantees significant hypotension:
Dosage: Some general concepts should be applied when choosing the dose of intravenous anesthetic. Administering a larger-than-required dose virtually guarantees significant hypotension: Elderly patients require lower doses compared with nonelderly adults, and children require higher doses (on a milligram-per-kilogram scale) compared with adults.
Elderly patients require lower doses compared with nonelderly adults, and children require higher doses (on a milligram-per-kilogram scale) compared with adults.

 Ketamine:
Ketamine: In contrast to GABA-mimetic drugs, ketamine:
In contrast to GABA-mimetic drugs, ketamine:• Is dysphoric: It produces very unpleasant sensations, hallucinations, and bad dreams. A benzodiazepine can be quite effective in reducing the dysphoria and is commonly coadministered with ketamine
 Propofol:
Propofol: Is a strong myocardial depressant and should be given very cautiously, if at all, in patients with a weak heart (low systolic function).
Is a strong myocardial depressant and should be given very cautiously, if at all, in patients with a weak heart (low systolic function).









Local Anesthetics




MOA (Mechanism of Action)
 Neuronal transmission requires that an action potential be propagated from one end of a neuron to the other. Voltage-gated sodium channels open up as the wave of depolarization travels from one end of the neuron to the other. The opening of these ion channels permits sodium to enter the cell, causing depolarization, and is the primary method by which the wave of depolarization occurs.
Neuronal transmission requires that an action potential be propagated from one end of a neuron to the other. Voltage-gated sodium channels open up as the wave of depolarization travels from one end of the neuron to the other. The opening of these ion channels permits sodium to enter the cell, causing depolarization, and is the primary method by which the wave of depolarization occurs. Local anesthetics bind these voltage-gated sodium channels. The ion channels can exist in three states: resting, activated (open), and inactivated. The local anesthetic binds them in the inactivated state and prevents them from transitioning to the open state; thus the ion channel remains closed and unresponsive to incoming depolarizing currents (Figure 21-4).
Local anesthetics bind these voltage-gated sodium channels. The ion channels can exist in three states: resting, activated (open), and inactivated. The local anesthetic binds them in the inactivated state and prevents them from transitioning to the open state; thus the ion channel remains closed and unresponsive to incoming depolarizing currents (Figure 21-4).




 Voltage-gated sodium channels are the primary ion channels in phase 0 of the cardiac action potential. Therefore, local anesthetics are also classified as antiarrhythmics, specifically type 1b. This mechanism of action is discussed in more detail in the discussion of Na+ channel blockers in Chapter 11 and is also responsible for cardiac toxicity of local anesthetics.
Voltage-gated sodium channels are the primary ion channels in phase 0 of the cardiac action potential. Therefore, local anesthetics are also classified as antiarrhythmics, specifically type 1b. This mechanism of action is discussed in more detail in the discussion of Na+ channel blockers in Chapter 11 and is also responsible for cardiac toxicity of local anesthetics.
Pharmacokinetics
 Local anesthetics are most commonly injected, but other routes of administration include topical application: oral sprays, creams, and vaporized forms (for airways).
Local anesthetics are most commonly injected, but other routes of administration include topical application: oral sprays, creams, and vaporized forms (for airways). There are important factors that dictate the potency, speed of onset, and duration of a local anesthetic:
There are important factors that dictate the potency, speed of onset, and duration of a local anesthetic: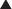 Lipid solubility: Increased lipid solubility results in more drug being able to cross the cell membrane and bind the ion channel. The property of increased lipid solubility influences:
Lipid solubility: Increased lipid solubility results in more drug being able to cross the cell membrane and bind the ion channel. The property of increased lipid solubility influences:

 Amide local anesthetics are metabolized (N-dealkylation and hydroxylation) by microsomal P-450 enzymes in the liver (Table 21-3).
Amide local anesthetics are metabolized (N-dealkylation and hydroxylation) by microsomal P-450 enzymes in the liver (Table 21-3).TABLE 21-3 Local Anesthetic Durations
| Agent | Duration Plain (minutes) | Duration with Epinephrine (minutes) |
|---|---|---|
| 2-Chloroprocaine | 20-30 | 30-45 |
| Procaine | 15-30 | 30 |
| Lidocaine | 30-60 | 120 |
| Mepivacaine | 45-90 | 120 |
| Prilocaine | 30-90 | 120 |
| Bupivacaine | 120-240 | 180-240 |



