Chapter 21 Neurological disorders – epilepsy, Parkinson’s disease and multiple sclerosis
• This chapter focuses on several common neurological disorders, each of which has a wide range of therapeutic strategies available. These disorders are: epilepsy, Parkinson’s disease and multiple sclerosis. The treatments of other common neurological disorders are covered in other sections: namely: headaches (Pain section: Ch. 18), stroke (Ch. 24) and dementia (Ch. 20).
• It also touches on the pharmacological principles of other neurological disorders, including: movement disorders other than Parkinson’s disease; spasticity (that is a physical sign characteristic of certain diseases such as stroke or multiple sclerosis); peripheral neuropathy; motor neurone disease; tetanus.
Epilepsy
Definitions
A seizure is a clinical symptom or sign caused by abnormal electrical discharges within the cerebral cortex.1 For example, a tonic–clonic seizure refers to a pattern by which a patient loses consciousness, becomes generally stiff (tonic), and subsequently jerks all limbs (clonus); whereas a complex partial seizure refers to a constellation of impaired consciousness, déjà vu sensations, epigastric rising sensation, olfactory hallucinations and motor automatisms, e.g. lip smacking.2 By contrast, epilepsy refers to the clinical syndrome of recurrent seizures, and implies a pathological state that predisposes to further future seizures. Hence having one, or even a single cluster of seizures (i.e. over a few days) does not in itself qualify as epilepsy, since these seizures may have been due to a febrile illness or drug intoxication that themselves later resolve. By contrast, having at least two seizures, separated by at least a few weeks, is usually sufficient to signify epilepsy. Only one-third of people having seizures develop chronic epilepsy.
Pathology and seizure types
Epilepsy affects 0.5–1% of the general population, while the lifetime risk of having a seizure is 3–5 %. There are both multiple causes and multiple seizure types.3 Approximately half of adult epilepsy is believed to be due to genetic or early developmental causes, although the exact nature of these – e.g. sodium channel mutations or cerebral palsy – are determined in only a small minority. The other half of adult epilepsy is due to acquired causes, such as alcohol, stroke, traumatic head injury or brain tumours.
The cause of epilepsy determines the seizure type. Genetic causes (i.e. ‘primary’) predispose to generalised seizures4 characterised by tonic–clonic or absence seizures (lapses of consciousness lasting seconds), myoclonus (random limb jerk at other times), photosensitivity (seizures triggered by flashing lights), EEG showing a 3-Hz spike-and-wave pattern, and a normal MRI brain. Conversely, where focal brain injury has occurred, e.g. brain tumour or stroke, and the brain scan is abnormal, focal epileptic discharges occur within the brain leading to a partial seizure – i.e. when only a narrow set of brain functions are disturbed, e.g. causing single limb jerking (implying motor cortex involvement). Importantly, partial seizures can propagate very quickly to become a ‘secondary generalised seizure’. Another common cause of adult-onset partial epilepsy is maldevelopment of the medial temporal lobes (‘mesial temporal sclerosis’) believed to be due to injury, e.g. hypoxia or infection, during fetal or early childhood life, and sometimes apparent as atrophic hippocampi and amygdala on high-resolution MRI.
Principles of management
• Identification of underlying cause and treatment of this where possible, e.g. cerebral neoplasm or arteriovenous malformation.
• Educate the patient about the disease, duration of treatment and need for compliance.
• Counselling the patient about avoiding harm from seizures, e.g. driving regulations, swimming or bathing alone and climbing, as well as other dangerous pursuits, to be avoided.
• Avoid precipitating factors, e.g. alcohol, sleep deprivation, stroboscopic light.
• Anticipate natural variation, e.g. fits may occur particularly or exclusively around menstruation in women (catamenial5 epilepsy).
• For most cases with recurrent seizures, an antiepileptic drug is prescribed with subsequent monitoring and adjustment of dosage or drug type (see below).
• Consider surgical therapies in patients with refractory seizures, e.g. vagal nerve stimulation, temporal lobectomy. For childhood refractory epilepsy, a ketogenic diet – i.e. high fat:carbohydrate ratio – is useful, as ketone bodies are antiepileptogenic.
• Acute treatment of generalised convulsive seizures consists of ensuring the patient lies on the floor away from danger, and is postictally manoeuvred into the recovery position. If a seizure continues for more than a few minutes, rectal or buccal diazepam or intranasal midazolam can be given. If convulsive seizures last for more than 5 min, patients should be transferred to hospital for consideration of intravenous benzodiazepine and phenytoin.
Practical guide to antiepilepsy drugs
1. When to initiate. Following a single seizure the chance of a further seizure is approximately 25% over the following 3 years. Furthermore, only 33% of single-seizure patients develop chronic epilepsy. Hence the majority of first seizures are provoked by a reversible, and often recognisable, factor, e.g. infection, drug toxicity, surgery. For these reasons, following a single seizure6 anticonvulsants are not generally prescribed, whereas after two or more distinct seizure episodes (i.e. with more than a few weeks apart between episodes), they generally are prescribed. Immediate treatment of single or infrequent seizures does not affect long-term remission but introduces the potential for adverse effects. Patients need to be made aware that anticonvulsant therapy reduces harm caused by generalised seizures, and may also reduce the risk of sudden death in epilepsy (SUDEP), that usually occurs during sleep.
2. Monotherapy. Although the choice of anticonvulsants is large (approximately 20), first–line therapy is generally restricted to one of only a few drugs that have a good track record and are relatively safe and well-tolerated. Initial therapy is confined to a single drug (i.e. monotherapy) that is usually effective in stopping seizures or at least significantly decreasing their frequency. The majority of epilepsy patients (70%) can remain on monotherapy for adequate control, although sometimes the choice of monotherapy may need to be switched to allow for tolerance or optimisation of seizure control. As the number of single anticonvulsants tried increases, the incremental likelihood that any new one will offer a significant reduction in seizures decreases: from 50% response to a first drug, to an additional 30% to a second drug, to an extra 10% to a third drug, and less than 5% for any subsequent drug tried.
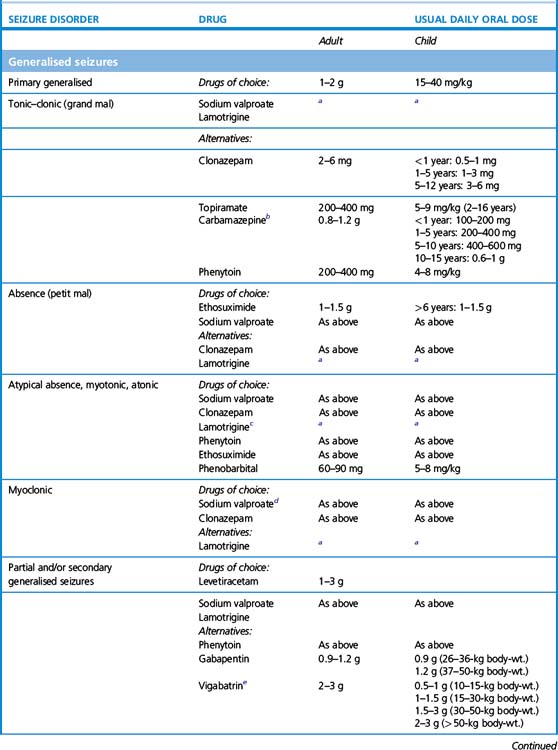
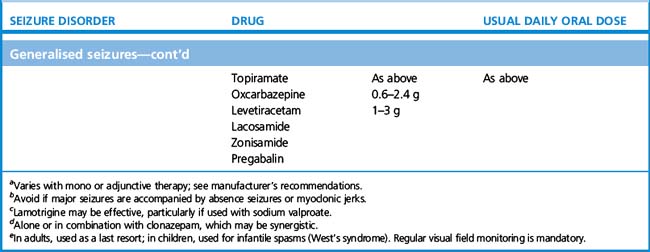
3. What drug to initiate. For older types of anticonvulsants, knowing the seizure type – i.e. whether partial or primary generalised – mattered, because in certain cases the spectrum of seizure efficacy is limited, and, moreover, certain seizure types can be worsened by ill-chosen drugs. For example, carbamazepine is an effective first-line therapy for partial seizures but may worsen primary generalised, absence or myoclonic seizures; similarly phenytoin can worsen absence and myoclonic seizures. Ethosuximide, by contrast, is only effective in primary generalised, and not partial, seizures.
More modern anticonvulsants, by contrast, are in general effective over a much broader range of seizure types allowing for more confidence of use even when seizure type is uncertain. Thus sodium valproate, lamotrigine and levetiracetam are active against both primary and secondary generalised epilepsy, and being relatively well tolerated, account for most first-line prescriptions. In one head-to-head study comparing popular first-line therapies for generalised and partial seizures, lamotrigine was generally tolerated better than other drugs, while valproate was the most efficacious; carbamazepine and topiramate were more likely to cause unwanted effects.7
4. Women of reproductive age and children. These categories of patients prompt selection of particular drugs and avoidance of others (see below for more detail).
5. Polytherapy. If a trial of three or so successive anticonvulsants (i.e. taken as monotherapy at adequate dosage for at least several months) does not control a patient’s epilepsy, it may be worthwhile trying dual therapy. Polytherapy offers the theoretical advantage of controlling neuronal hyperexcitability by more than one mechanism, that can be synergistic. In reality, increasing polytherapy often adheres to the law of diminishing returns, viz. the proportion of uncontrolled patients who show a positive response decreases at each addition of drug number And at the same time, adverse effects become more likely.
6. Abrupt withdrawal. Effective therapy must never be stopped suddenly, as this is a well-recognised trigger for status epilepticus, which may be fatal. But if rapid withdrawal is required by the occurrence of toxicity, e.g. due to a severe rash or significant liver dysfunction, a new drug ought to be started simultaneously. The speed by which the dose of a new drug can be raised varies according to drug type and urgency.
7. Circumstantial seizures. In cases where fits are liable to occur at a particular time, e.g. the menstrual period, adjust the dose to achieve maximal drug effect at this time or confine drug treatment to this time. For example, in catamenial epilepsy, clobazam can be useful given only at period time.
Failure to respond
• Non-compliance, diarrhoea and vomiting, patients instructed to be ‘nil by mouth’ (revealed by measuring blood concentrations of drug).
• Inadequate dosing, including the possibility of drug interaction, e.g. another drug reducing the effective dose of the anticonvulsant by hepatic enzyme induction.
• Pregnancy also causes hepatic induction, and reduces the effective dose of lamotrigine.
• Increase in the severity of an underlying disease, e.g. enlargement of a brain tumour, or new disease.
• Drug resistance, e.g. genetic polymorphisms in hepatic cytochromes (such as CYP 2 C9) that metabolise drugs, sodium channel subunit SCN1A, or the P glycoprotein drug transporter (ABCB1 gene) that expels drugs from neurones.
Drug withdrawal
If patients have remained seizure-free for more than a few years, it is reasonable to consider withdrawal of antiepilepsy drug therapy.8,9 The prognosis of a seizure disorder is determined by:
• Type of seizure disorder – benign rolandic epilepsy, solely petit mal or grand mal seizures confer a high chance of full remission, whereas juvenile myoclonic epilepsy, temporal or frontal lobe epilepsies often require lifelong treatment.
• Time to remission – early remission carries a better outlook.
• Number of drugs required to induce remission – rapid remission on a single drug is a favourable indicator for successful withdrawal.
• MRI brain scan findings – presence of an underlying lesion predicts difficult control.
• EEG findings – epileptogenic activity is a predictor of poor outcome for drug withdrawal.
• Associated neurological deficit or learning difficulty – control is often difficult.
• Length of time of seizure freedom on treatment – the longer the period, the better the outlook.
Pregnancy and epilepsy
Pregnancy worsens epilepsy in about a third of patients, but also improves epilepsy in another third. One of the main concerns in this patient group is that all anticonvulsants increase the chance of teratogenicity slightly, with valproate, phenytoin and phenobarbital carrying most risk. The toxicological hazard must be weighed against the risk of seizures which themselves can be harmful to mother and unborn baby, and are likely to worsen if anticonvulsants are discontinued. For instance, the risk of major congenital anomalies in the fetus is 1% for healthy mothers, 2% in untreated epileptic mothers (in observational studies, so generally not severe epileptics), and 2–3% in mothers on epilepsy monotherapy. Valproate, by contrast, has been associated with a malformation rate of approximately 10%,10 while 20–30% of children are subsequently found to have mild learning disabilities or require ‘special needs’ education. The UK maintains a national drug monitoring register of all pregnant women taking antiepileptic drugs.
• neural tube defects are related to deficiencies in folic acid stores before pregnancy, so that antiepileptic drugs that affect stores, e.g. valproate, can be avoided, and folic acid 5 mg per day given for several months in advance, and
• adjustments in dose and type of drug can be avoided in the early stages of pregnancy as there is a higher risk of toxicity and seizure breakthrough during this critical phase of fetal development. In general, patients having seizures with blackouts should be on an effective dose of an anticonvulsant, because of the risks of anoxia, lactic acidosis and trauma.
Status epilepticus
Always investigate and treat the cause of a generalised seizure. Give aciclovir i.v. if viral encephalitis is suspected or, if status is triggered by removing an antiepileptic drug, it must be re-instituted. Magnesium sulphate is the treatment of choice for seizures related to eclampsia (see also p. 125).11
Details of further management appear in Table 21.1.
Table 21.1 Treatment of status epilepticus in adults
| Status | Treatment |
|---|---|
| Early | Lorazepam 4 mg i.v., repeat once after 10 min if necessary, or clonazepam 1 mg i.v. over 30 s, repeat if necessary, or diazepam 10–20 mg over 2–4 min, repeat once after 30 min if necessary |
| Established | Phenytoin 15–18 mg/kg i.v. at a rate of 50 mg/min, and/or phenobarbital 10–20 mg/kg i.v. at a rate of 100 mg/min |
| Refractory | Thiopental or propofol or midazolam with full intensive care support |
Pharmacology of individual drugs
Modes of action
Enhancement of gamma-aminobutyric acid (GABA) transmission
Examples: benzodiazepines, phenobarbital, valproate, vigabatrin, tiagabine.12 By enhancing GABA, the principal inhibitory transmitter of the brain, neuronal membrane permeability to chloride ions is increased, which secondarily reduces cell excitability. Benzodiazepines and barbiturates activate the GABA receptor via specific benzodiazepine and barbiturate binding sites.