DIAGNOSIS
Headaches are divided into primary and secondary types.
- Primary headache syndromes are not associated with other diseases and include migraine, tension-type, and cluster headaches.
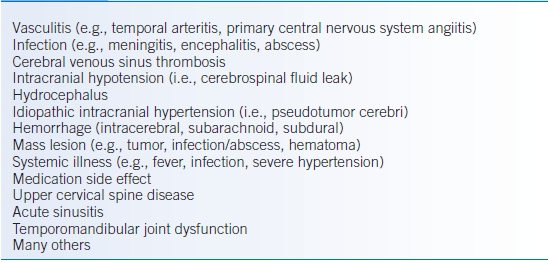
- Secondary headaches are common manifestations of other diseases (Table 43-2).
TABLE 43-2 Secondary Headache Differential Diagnosis
Clinical Presentation
- Headache diagnosis relies on a detailed medical history (e.g., frequency, duration, location, severity, photophobia, phonophobia, nausea, vomiting, autonomic signs).
- Red flags (Table 43-1) increase the suspicion for secondary headache disorders.
- Abnormal neurologic signs suggest secondary headache disorders.
- General and neurologic examination may include funduscopy, palpation of temporal arteries, auscultation for carotid bruits, palpation of the temporomandibular joint, and examination of neck and shoulder muscles.
Diagnostic Criteria
Migraine
- Divided into two major subtypes:
- Without aura (common migraine). See Table 43-3 for detailed diagnostic criteria.4
- With aura (classical migraine; approximately one-third of patients).
- Without aura (common migraine). See Table 43-3 for detailed diagnostic criteria.4
- Auras are recurrent attacks of reversible focal neurologic symptoms that develop over 5 to 20 minutes, last for <60 minutes, and are often associated with a migraine headache.4
- Visual auras are most common and can include fortification spectra and scotoma.
- Less common aura symptoms may include paresthesias, numbness, weakness, gait instability, speech change, and others.
- Visual auras are most common and can include fortification spectra and scotoma.
TABLE 43-3 Migraine Without Aura Diagnostic Criteria

Data from Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004;24:9–160.
Tension-Type Headache
- Most common type of primary headache.
- See diagnostic criteria for tension-type headaches in Table 43-4.4
TABLE 43-4 Tension-Type Headache Diagnostic Criteria

Data from Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004;24:9–160.
Cluster Headache
- Most common trigeminal autonomic headache characterized by brief (15 to 180 minutes), severe, unilateral pain. See diagnostic criteria in Table 43-5.4
- Cluster headaches are more common in men than in women.
- Unlike migraine, patients are often restless during cluster headaches.
TABLE 43-5 Cluster Headache Diagnostic Criteria

Data from Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004;24:9–160.
Medication Overuse Headache
- A secondary headache type defined as an interaction between a medication used excessively in a susceptible patient.4
- Most common cause of headache occurring on more than 15 days/month in the setting of analgesic overuse (>15 days/month for >3 months).4
- Can develop with analgesic overuse in patients with tension, migraine, or cluster headaches.
- Acetaminophen, aspirin, opiates, and combinations of caffeine and butalbital (e.g., Fiorinal, Fioricet, and Esgic) are frequently implicated.
Diagnostic Testing
- No testing is warranted with a typical history, no red flags, and a normal examination.
- Testing may include the following if history and exam suggest secondary headache:
- Head CT
- MRI of the brain with and without contrast
- Angiography (magnetic resonance angiography [MRA], computed tomography angiography [CTA], carotid ultrasound, or conventional catheter)
- Blood tests (e.g., erythrocyte sedimentation rate [ESR])
- Cervical spine imaging
- Lumbar puncture (LP)
- Head CT
TREATMENT
- Patient and family education
- The most effective treatment for headaches is prevention.
- Headache hygiene: recognition and avoidance of migraine triggers including emotional stress, hormonal fluctuation, missed meals, caffeine withdrawal, weather changes, sleep disturbance, muscular tension, alcohol, heat, dehydration, and certain foods. A headache diary may be helpful in elucidating triggers in individual patients.
- Discuss limiting acute treatments to minimize medication overuse headaches.
- The most effective treatment for headaches is prevention.
- Stop analgesic overuse
- Cessation of the offending acute medications may be difficult for many patients.
- Headache transformation is likely to occur with 5 days of butalbital, 8 days of opioid, 10 days of triptan, and 10 to 15 days of NSAID use per month.5
- Acute migraine treatment should be limited to 2 or fewer days per week.
- Opioids and butalbital should be avoided.
- Headache transformation is likely to occur with 5 days of butalbital, 8 days of opioid, 10 days of triptan, and 10 to 15 days of NSAID use per month.5
- Following cessation, headaches may temporarily get worse.
- Multiple strategies can be employed based on individual circumstances: stop or taper acute medications, steroid taper, hospitalization for rescue medication (e.g., dihydroergotamine [DHE], prochlorperazine, droperidol), and/or initiation of prophylactic medication.
- Cessation of the offending acute medications may be difficult for many patients.
Medications
Acute Headache Treatment (Migraine and Tension Type)
- The following medications used to treat acute headaches are most effective when taken at headache onset and may be used in combination.
- Use of butalbital-containing medications (e.g., Fioricet, Fiorinal, and Esgic) should be approached with caution.
- Over-the-counter (OTC) analgesics: NSAIDs, acetaminophen, aspirin, and caffeine are often effective as a first-line therapy and may be combined for better effect.
- Antiemetics:
- Commonly used to treat migraine pain, particularly if headaches are accompanied by nausea/vomiting.
- Examples include metoclopramide (Reglan) 10 mg IV and prochlorperazine (Compazine) 10 mg IV/IM/PR.6,7 Droperidol has also been shown to be effective; however, there is an FDA boxed warning regarding QT prolongation and torsades de pointes with IV administration. A 12-lead ECG must be obtained prior to administration of droperidol and the patient monitored for arrhythmias afterwards. Promethazine (Phenergan) should not be given IV/SC due to the risk of severe tissue necrosis/gangrene with extravasation (FDA boxed warning).
- Side effects include sedation, akathisia, and QT prolongation. Diphenhydramine (12.5 mg IV) may be given concomitantly to prevent akathisia and dystonic reactions.
- Commonly used to treat migraine pain, particularly if headaches are accompanied by nausea/vomiting.
- Triptans
- Triptans are serotonin receptor 1b/1d agonists that are thought to be relatively specific in their effectiveness for migraine headache. Multiple studies have demonstrated effectiveness.8 All are most effective when used early.
- Multiple triptans exist (e.g., almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, and zolmitriptan) and differ in half-life, route of administration, and cost. Common regimens include the following:
- Sumatriptan 25 to 100 mg PO ×1, may repeat after 2 hours, maximum dose 200 mg/24 hours.9 Sumatriptan 4 to 6 mg SC ×1, may repeat after 1 hour, maximum dose 12 mg/24 hours.10 SC administration is faster in onset but associated with more side effects. Sumatriptan 5 to 20 mg intranasal ×1, may repeat after 2 hours, maximum dose 40 mg/24 hours.11
- Eletriptan 20 to 40 mg ×1, may repeat after 2 hours, maximum dose 80 mg/24 hours.
- Rizatriptan 5 to 10 mg ×1, may repeat after 2 hours, maximum dose 30 mg/24 hours.
- Almotriptan 6.25 to 12.5 mg ×1, may repeat after 2 hours, maximum dose 25 mg/24 hours.
- Zolmitriptan 1.25 to 2.5 mg PO ×1, may repeat after 2 hours, maximum dose 10 mg/24 hours. Zolmitriptan 2.5 to 5 mg intranasal ×1, maximum dose 10 mg/24 hours.
- Sumatriptan 25 to 100 mg PO ×1, may repeat after 2 hours, maximum dose 200 mg/24 hours.9 Sumatriptan 4 to 6 mg SC ×1, may repeat after 1 hour, maximum dose 12 mg/24 hours.10 SC administration is faster in onset but associated with more side effects. Sumatriptan 5 to 20 mg intranasal ×1, may repeat after 2 hours, maximum dose 40 mg/24 hours.11
- All available oral triptans appear to be relatively equally effective.12
- Contraindications exist due to the vasoconstrictive effects and include ischemic heart disease, coronary artery vasospasm, peripheral arterial disease, uncontrolled hypertension, headaches associated with weakness, and concurrent (or within 2 weeks) monoamine oxidase inhibitor (MAOI) use.
- Monitor for symptoms of serotonin syndrome, which could arise with monotherapy or coadministration with a selective serotonin reuptake inhibitor (SSRI) or serotonin norepinephrine reuptake inhibitor (SNRI), necessitating discontinuation of drug and supportive care.
- Triptans are serotonin receptor 1b/1d agonists that are thought to be relatively specific in their effectiveness for migraine headache. Multiple studies have demonstrated effectiveness.8 All are most effective when used early.
- Dihydroergotamine
- DHE is an agonist of multiple serotonin receptor subtypes, including 1b/1d, dopamine receptors, and is also a nonselective α-adrenergic blocker. It may be used for acute migraine treatment and is often used to break status migrainosus (debilitating migraine lasting >72 hours).
- Alone, DHE may be less effective than a triptan or an antiemetic, but when combined with an antiemetic it is superior to ketorolac and meperidine. DHE may also be more effective at preventing recurrences.8,13
- Parenteral DHE is usually given in combination with an IV antiemetic (prochlorperazine or metoclopramide) due to the fairly high rate of DHE-induced nausea.
- For acute migraine treatment
- DHE 1 mg IM or SC, may repeat q1 hour to a maximum dose of 3 mg/day.
- DHE 1 mg IV, may repeat q1 hour to a maximum dose of 2 mg/day.
- DHE nasal 0.5 mg (1 spray) each nostril, may repeat in 15 minutes, maximum 2 mg (4 sprays)/attack or 3 mg (6 sprays)/day.
- DHE 1 mg IM or SC, may repeat q1 hour to a maximum dose of 3 mg/day.
- DHE for intractable migraine is an off-label usage but has been found to be effective if several trials.14–16 The typical protocol starts with IV metoclopramide 10 mg over 30 minutes. A 0.5 test dose of DHE is then given. Those with persistent headache are given 1 mg DHE IV with 10 mg metoclopramide every 8 hours, typically over 2 to 3 days.
- Contraindications are similar to that of triptans. Serotonin syndrome may occur when DHE is administered with an SSRI. Coadministration with CYP3A4 inhibitors (e.g., protease inhibitors, macrolides, and azole antifungals) is also contraindicated.
- DHE is an agonist of multiple serotonin receptor subtypes, including 1b/1d, dopamine receptors, and is also a nonselective α-adrenergic blocker. It may be used for acute migraine treatment and is often used to break status migrainosus (debilitating migraine lasting >72 hours).
Prophylactic Treatment (Migraine and Tension Type)
- The decision to treat with prophylactic medications is based upon the frequency and duration of headaches, amount of disability caused, and response to acute headache medicines.17
- Nonpharmacologic therapies include physical therapy and behavioral therapy (e.g., biofeedback and relaxation training).
- Anticonvulsants17–19:
- Topiramate: start at 25 mg daily and increase to 50 mg bid, maximum dose 200 mg/day.20 Central nervous system (CNS) side effects are relatively common including paresthesia, fatigue, drowsiness, dizziness, nervousness, memory and language difficulty, and poor concentration. Anorexia, nausea, dysgeusia, diarrhea, and weight loss are also fairly common. Side effects are more common with higher doses.
- Valproic acid/divalproex: start at 250 mg bid and increase to a maximum of 500 mg bid.21 Extended release divalproex may be given 200 to 1,000 mg daily. There is an FDA boxed warning for hepatotoxicity and teratogenicity. Common side effects include nausea/vomiting, weight changes, hair loss, drowsiness, insomnia, tremor, and thrombocytopenia.
- Gabapentin is most likely not effective.22
- Topiramate: start at 25 mg daily and increase to 50 mg bid, maximum dose 200 mg/day.20 Central nervous system (CNS) side effects are relatively common including paresthesia, fatigue, drowsiness, dizziness, nervousness, memory and language difficulty, and poor concentration. Anorexia, nausea, dysgeusia, diarrhea, and weight loss are also fairly common. Side effects are more common with higher doses.
- Antihypertensives17–19
- β-Blockers
- Propranolol: start at 80 mg/day divided tid/qid, increase to a maximum of 160 to 240 mg divided tid/qid, or use long-acting propranolol.
- Metoprolol: start at 50 mg bid, may increase to a maximum of 200 mg/day.
- Propranolol: start at 80 mg/day divided tid/qid, increase to a maximum of 160 to 240 mg divided tid/qid, or use long-acting propranolol.
- Calcium channel blockers do not have nearly as convincing evidence for migraine prophylaxis, but they are frequently used. Verapamil is most common, starting at 120 mg divided tid increased to a maximum of 480 mg divided tid, or extended release verapamil.
- Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARB) may also have some prophylactic efficacy.
- β-Blockers
- Antidepressants17–19
- Tricyclic antidepressants (TCAs): amitriptyline 10 to 150 mg at bedtime.23 Common side effects include dry mouth, drowsiness, and weight gain.
- SSRIs and SNRIs may also be effective. A small randomized trial showed that venlafaxine XR 150 mg was effective for migraine prophylaxis.24
- Tricyclic antidepressants (TCAs): amitriptyline 10 to 150 mg at bedtime.23 Common side effects include dry mouth, drowsiness, and weight gain.
- NSAIDs such as ibuprofen and naproxen may be beneficial for migraine prophylaxis but they can also be medication overuse headaches.
- OTC agents: probably/possibly effective agents include butterbur (Petasites hybridus) extract 50 to 75 mg bid, riboflavin (200 mg bid), magnesium (300 to 600 mg/day), feverfew extract (MIG-99) 6.25 mg tid, and Co-Q10 (100 mg tid).17,19,25
- Botulinum toxin is probably ineffective for intermittent migraine prophylaxis.19 It may, however, be effective for chronic, refractory headaches.26
Cluster Headache Treatment
- Prevention includes avoidance of triggers including alcohol and nitrates.
- Abortive therapies include inhaled oxygen by mask (high flow), triptans (SC or nasal), and DHE (see above).16,27,28
- Prophylaxis can be attempted with verapamil, lithium, valproate, corticosteroids, and melatonin.29
Seizures
GENERAL PRINCIPLES
- A seizure is a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.30
- Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures.30 The occurrence of a single seizure does not necessitate the diagnosis of epilepsy.
- An unprovoked seizure occurs without a precipitant, thus excluding those associated with acute CNS insult or metabolic/toxic disturbance including alcohol withdrawal.
- Seizures affect almost 2 million Americans. The age-adjusted prevalence of epilepsy is 6.8/1,000 population.31 The cumulative incidence of having an unprovoked seizure is 4.1% and the cumulative incidence of epilepsy is 3.1%.32
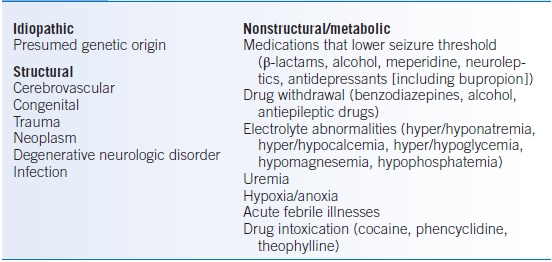
- Table 43-6 presents a list of the more common causes of seizures.
- Focal seizures arise from epileptogenic foci in a localized region of one cerebral hemisphere and are further classified based upon level of consciousness.33 Focal seizures include simple partial seizures and complex partial seizures.
- Generalized seizures involve widespread regions in both brain hemispheres with impairment of consciousness and include nonconvulsive (absence) and convulsive (myoclonic, clonic, tonic, tonic-clonic, and atonic) seizures.
- Simple partial seizures
- Involve no impairment of consciousness.
- Usually brief, lasting approximately 15 seconds to 2 minutes.
- Can manifest as stereotypic motor movements, abnormal somatosensory symptoms (e.g., paresthesias), special sensory symptoms (e.g., visual, auditory, olfactory, gustatory, or vertigo), neuropsychiatric symptoms (e.g., emotion, memory, cognition, or perceptions), and autonomic symptoms (e.g., epigastric rising, pallor, flushing, or piloerection).
- Involve no impairment of consciousness.
- Complex partial seizures
- Stereotyped partial seizures with impaired consciousness.
- May include motionless staring, behavioral arrest, unresponsiveness, oral or limb automatisms, focal limb posturing, or clonus.
- Usually last 30 seconds to 3 minutes and are often followed by postictal confusion.
- Stereotyped partial seizures with impaired consciousness.
- Secondarily generalized tonic-clonic seizures
- Secondary generalization is caused by the spread of ictal discharge from a localized focus to involve both hemispheres symmetrically.
- Tonic and/or clonic movements are usually asymmetric.
- Often accompanied by postictal confusion.
- Secondary generalization is caused by the spread of ictal discharge from a localized focus to involve both hemispheres symmetrically.
- Typical absence seizure
- Characterized by sudden behavioral arrest, loss of awareness, and blank staring.
- Brief, lasting 5 to 30 seconds with immediate return to baseline consciousness.
- Characterized by sudden behavioral arrest, loss of awareness, and blank staring.
- Generalized tonic-clonic seizures
- Generalized tonic-clonic seizures are characterized by tonic stiffening of axial and limb muscles lasting 10 to 15 seconds.
- The clonic phase involves clonic jerking that increases in frequency and amplitude with bilateral upper extremity flexion and bilateral lower extremity extension.
- Tonic and/or clonic movements are usually symmetric and last 1 to 2 minutes.
- Consciousness recovers in minutes but postictal confusion may last for hours.
- Generalized tonic-clonic seizures are characterized by tonic stiffening of axial and limb muscles lasting 10 to 15 seconds.
- Psychogenic nonepileptic seizures (PNES)
- PNESs are paroxysmal changes in behavior that superficially resemble epilepsy but have a psychological rather than a medical cause.34
- 12% to 20% of patients seen in epilepsy clinics have PNES but 10% to 30% of PNES patients also have epilepsy.
- Treatment consists of psychological intervention and weaning off antiepileptic medications.
- PNESs are paroxysmal changes in behavior that superficially resemble epilepsy but have a psychological rather than a medical cause.34
TABLE 43-6 Etiology of Seizures
DIAGNOSIS
Clinical Presentation
- The goal of the history is to identify possible triggers of provoked seizures and to distinguish seizures from other paroxysmal events, such as syncope, episodic movement disorders, PNES, narcolepsy/cataplexy, and transient ischemic attacks (TIAs).
- A reliable description of the seizure should be obtained (preferably from a bystander when consciousness is impaired). However, bystander reports of seizure duration are often unreliable.
- The purpose of a detailed neurologic examination is to elicit focal findings suggestive of a structural lesion.
Diagnostic Testing
- The purpose of diagnostic testing is to distinguish provoked seizure from unprovoked seizure, and to determine the risk of recurrence.
- Electroencephalogram (EEG)
- A normal EEG does not rule out epilepsy.
- 30% to 40% of first EEGs show abnormalities. Early EEGs within 24 hours have a higher yield (51%) than after 24 hours (34%).35
- Epileptiform abnormalities may confirm a diagnosis of epileptic seizures and distinguish partial seizures from generalized seizures.
- An abnormal EEG approximately doubles the risk of seizure recurrence.36
- Extended video-EEG (e.g., 24-hour monitoring) is helpful in evaluating seizure types (including PNES) and in localizing seizure origins for possible surgical intervention.
- Ambulatory EEG is often less useful due to artifacts in the record and difficulty correlating unmonitored clinical events with the EEG tracing.
- A normal EEG does not rule out epilepsy.
- Brain MRI: workup of a seizure should include brain MRI with and without contrast to evaluate for structural lesions (e.g., mesial temporal sclerosis) that may benefit from surgical resection.
- LP: used to rule out CNS infections or carcinomatosis if there is clinical suspicion.
TREATMENT
- First seizures generally do not require antiepileptic drug (AED) treatment.
- Recurrence risk for a second unprovoked seizure is 34% at 5 years.
- The probability of achieving seizure control with AED treatment is the same for those treated immediately versus those treated after the second seizure.37
- Recurrence risk for a second unprovoked seizure is 34% at 5 years.
- The goal for the management of epilepsy is to minimize seizure frequency and medication side effects.
- Titrate medication dosage to find the lowest dose necessary to control seizures.
- Decrease modifiable triggers such as sleep deprivation, alcohol intake, caffeine, and stress.
- Implement seizure calendars to monitor seizure frequency.
- Titrate medication dosage to find the lowest dose necessary to control seizures.
- Patients with intractable epilepsy, despite sufficient trials of multiple AEDs, may be candidates for resective epilepsy surgery and should be referred to an epilepsy center.
Medications
First-Generation AEDs
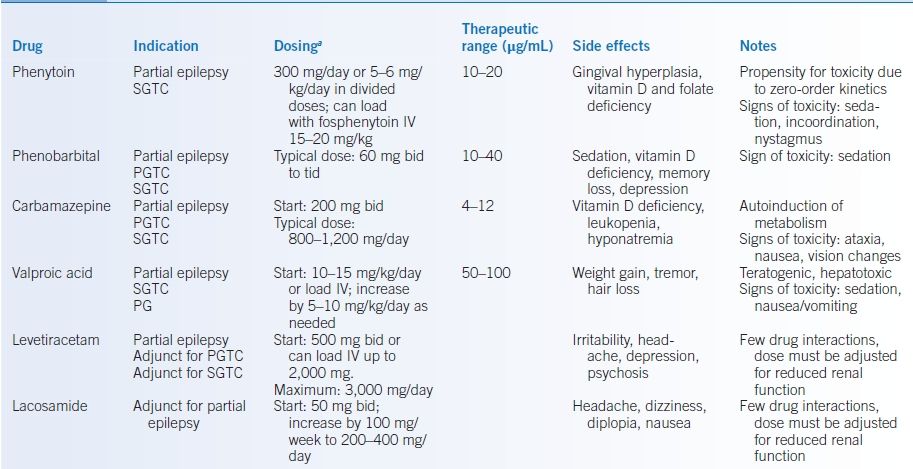
- These include phenytoin, carbamazepine, phenobarbital, and valproic acid (see Table 43-7).
- Advantages of use include ease of monitoring therapeutic serum levels.
- Carbamazepine has been associated with an increased risk of Stevens-Johnson in certain Asian populations carrying the HLA-B*1502 allele.
- Valproic acid can increase free phenytoin levels and inhibit the metabolism of other drugs.
- Drug-drug interactions of enzyme-inducing first generation AEDs (carbamazepine, phenytoin, and phenobarbital) are common, which result in the following:
- Increased metabolism of oral contraceptive pills and reduced hormone levels
- Increased metabolism of warfarin and reduced anticoagulant activity
- Increased TCA levels while TCAs increase AED levels
- Decreased levels of valproic acid
- Increased metabolism of oral contraceptive pills and reduced hormone levels
- Enzyme-inducing first-generation agents cause vitamin D deficiency. Supplementation with calcium and vitamin D helps prevent osteopenia and osteoporosis.
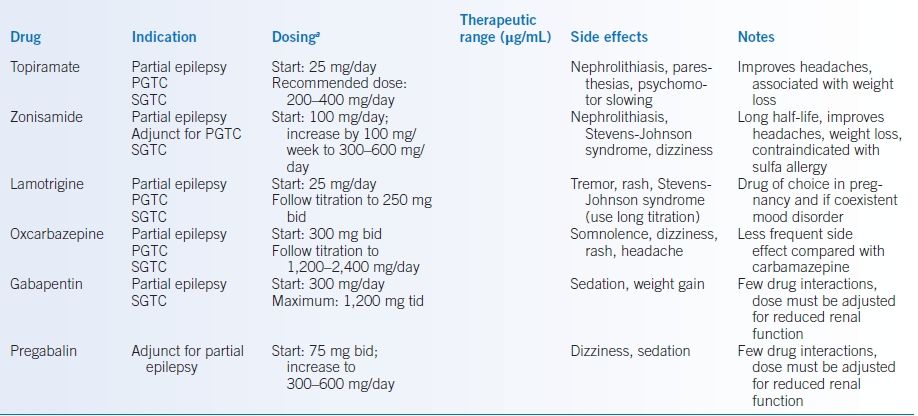
TABLE 43-7 Antiepileptic Drugs
aThese doses only represent guidelines and actual dosing may require lower or higher doses.
PG, primary generalized; PGTC, primary generalized tonic-clonic; SGTC, secondary generalized tonic-clonic.
Second-Generation AEDs
- These include levetiracetam, topiramate, zonisamide, lamotrigine, oxcarbazepine, gabapentin, pregabalin, and lacosamide (Table 43-7).
- Generally, they have better side-effect profiles, little to no need for serum monitoring, less frequent dosing, and fewer drug interactions.
- Rashes with mucosal involvement and systemic symptoms require immediate attention, as the rashes can represent potentially fatal reactions, such as toxic epidermal necrolysis or Stevens-Johnson syndrome.38
Benzodiazepines
- Used in the acute treatment of seizures; typically lorazepam 1 to 2 mg intramuscularly or intravenously, repeated as needed every 3 to 5 minutes while carefully monitoring for respiratory depression.
- Clonazepam is occasionally used long-term in patients with generalized epilepsy, particularly myoclonus, that is, refractory to multiple other medications. However, tolerance can develop necessitating escalating doses for seizure control.
- Side effects include sedation, irritability, ataxia, and respiratory depression.
Discontinuation of AEDs
- Unless specified by state laws or guidelines, physicians need to come to an agreement with their patients regarding prudent seizure precautions during the withdrawal period.
- Recurrence of seizures is unpredictable and influenced by the duration of active disease and seizure-free period.
- Tapering of AEDs should be slow (over 2 to 3 months) to avoid triggering a seizure.
- Taper one drug at a time for patients on polytherapy.
SPECIAL CONSIDERATIONS
Driving
- Longer seizure-free intervals (>6 to 12 months) are associated with reduced risk of motor vehicle accidents.
- Each state determines the required seizure-free interval, requirements for physician reporting, liability for driving recommendations, and whether mitigating factors are considered. Rules can be found at the Epilepsy Foundation website: http://www.epilepsy.com/learn/seizures-youth/about-teens-epilepsy/driving-and-transportation (last accessed 1/29/15).
Pregnancy
- All women of childbearing age on AEDs should be supplemented with folic acid 1 mg daily or 4 mg/day if taking valproic acid or carbamazepine.
- All AEDs are associated with teratogenic potential. Valproic acid is most associated with teratogenicity in the first trimester.39
Alcohol Withdrawal Seizures
- Alcohol withdrawal seizures are associated with relative or absolute withdrawal of ethanol.40
- They typically occur between 7 and 48 hours after cessation of drinking (peak 12 to 24 hours).
- Seizures are usually generalized tonic-clonic.
- Most patients (>60%) experience more than one seizure, and about 33% go on to develop delirium tremens, which continues to have a relatively high in-hospital mortality.
- Treatment involves IV benzodiazepines or the resumption of alcohol. Administer thiamine prior to providing glucose-containing fluids.
- There is no role for long-term AEDs unless there is an underlying epileptic disorder.
Multiple Sclerosis
GENERAL PRINCIPLES
- The incidence of MS is influenced by environmental and genetic factors. Prevalence in the United States is approximately 1/1,000. This increases to 1% with a first-degree relative, 5% in dizygotic twins, and 25% in monozygotic twins.
- Typically, multiple sclerosis (MS) is diagnosed in early adulthood and is more common in women (3:1).
- Patients may present with a relapsing or progressive course but must demonstrate dissemination in time and space.
- MS is an autoimmune-mediated process characterized by demyelinating plaques with predilection for periventricular white matter, corpus callosum, optic nerves, brainstem, cerebellum, and cervical spinal cord white matter.41 Gray matter (cortical) lesions are increasingly identified, but are poorly visualized on MRI.42
DIAGNOSIS
Clinical Presentation
- Relapsing-remitting MS (RRMS): 85% of MS patients begin with relapses (also called attacks or exacerbations) of worsening neurologic functioning followed by periods with partial or complete recovery.
- Secondary progressive MS (SPMS): 50% of RRMS patients may transform to a SPMS stage where deficits gradually worsen in-between relapses.
- Primary progressive MS (PPMS): patients have a gradual progressive disease course from onset with no specific relapses.
- Attacks typically evolve over a period of days, plateau, and then improve over weeks.
- Patients may present with a myriad of symptoms and signs including focal weakness, focal sensory complaints, Lhermitte sign (electric shock sensation down trunk and limbs induced by neck flexion), fatigue, unilateral loss of vision, diplopia, ataxia, or bladder complaints.
- A detailed neurologic examination is important to identify deficits related to demyelinating plaques and to monitor progression of disease.
Diagnostic Testing
- McDonald diagnostic criteria (2010 revision) are used for diagnosis to establish dissemination in time and space (see Table 43-8).43
- Brain and/or cervical spine MRI is used to identify and monitor lesions especially since some brain lesions can be clinically silent.
- Cerebrospinal fluid (CSF) often shows intrathecal antibody production (requires concurrent serum sample). Elevated CSF restricted oligoclonal bands are identified in >95% of MS patients.
- Visual-evoked potentials may identify optic nerve lesions.
- Other conditions may mimic MS and depending on the associated symptoms, it may be useful to check vitamin B12, thyroid-stimulating hormone (TSH), serum rapid plasma reagin (RPR), HIV, antinuclear antibodies (ANA), and extractable nuclear antigens (ENA).
- Evaluation for neuromyelitis optica (NMO) with an NMO-IgG should be considered if patient presents with recurrent optic neuritis and longitudinally extensive transverse myelitis on MRI (>3 vertebral segments).44 One-third of NMO patients are seronegative.
- Infections and metabolic disturbances must be considered as they can mimic relapses and cause pseudoexacerbations.
TABLE 43-8 Diagnostic Criteria for Multiple Sclerosis (2010 Revised McDonald Criteria)
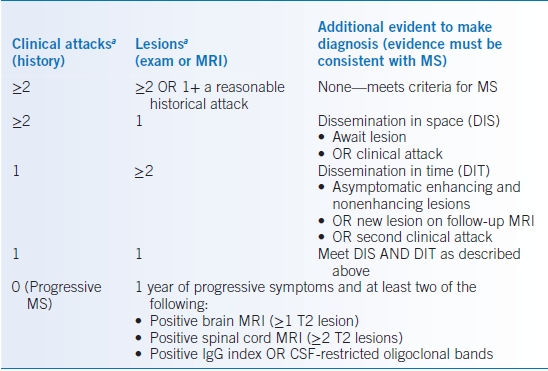
aDIS requires lesions in at least two of the following four CNS areas: periventricular, juxtacortical, infratentorial, and spinal cord.
Data from Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302.
TREATMENT
Acute Treatment
- IV methylprednisolone has been shown to hasten return to neurologic baseline. Typical dosing is 1 g daily, which can be divided, for 3 to 5 days. IV steroids may be followed by an oral prednisone taper over 4 weeks.45,46
- Plasma exchange is an option for severe relapses or if unresponsive/intolerant to steroids.47
- Physical therapy and occupational therapy can help address functional needs.
Disease-Modifying Therapy
- Consider referral to neurologist or dedicated MS center for long-term management.
- Immunomodulatory treatments aim to prevent further relapses, but do little to repair existing damage.
- Treatment of a clinically isolated syndrome, or a single monophasic attack, with immunomodulatory therapy may delay progression to clinically definite MS.48,49
- FDA-approved treatments for RRMS include interferon-β, glatiramer acetate, fingolimod, teriflunomide (active compound of leflunomide), and natalizumab.50
- Many of these medications (with the exception of glatiramer acetate) can cause leukopenias and liver enzyme abnormalities that must be monitored.
- These medications should not be used during pregnancy, although glatiramer acetate appears to be safe and is pregnancy category B.
- Many of these medications (with the exception of glatiramer acetate) can cause leukopenias and liver enzyme abnormalities that must be monitored.
- Natalizumab is a monoclonal antibody, which has been associated with progressive multifocal leukoencephalopathy (PML). Risk factors include length of treatment, prior treatment with immunosuppressant medications, and prior exposure to the JC virus.51
- There are no FDA-approved treatments for PPMS, although some patients may benefit from treatment particularly if they are younger or an MRI shows new lesions.52
Treatment of Related Symptoms
- Depression is common and is responsive to SSRIs or bupropion.
- Pain and spasms are common and may respond to medications, such as gabapentin, carbamazepine, duloxetine, tricyclics, and muscle relaxants.
- Urinary symptoms (spasticity, incontinence) may improve with treatment and warrant consideration of urologic consultation.
- Fatigue may respond to amantadine or modafinil.
Neuropathy
GENERAL PRINCIPLES
- Diabetes is the most common etiology of neuropathy in the US, while leprosy is the most common cause in undeveloped countries.
- Characterization of neuropathy is crucial to guide the differential diagnosis and includes temporal profile (onset and duration), family history, and anatomic classification including
- Fiber type (motor vs. sensory, large vs. small, somatic vs. autonomic)
- Axonal versus demyelinating
- Distribution affected (e.g., length dependent, specific dermatome, multifocal)
- Fiber type (motor vs. sensory, large vs. small, somatic vs. autonomic)
DIAGNOSIS
CLINICAL PRESENTATION
History
- Common complaints are paresthesias, numbness, pain, weakness, and autonomic symptoms.
- Questions should focus on duration, course, and distribution of complaints.
- Consider relationship to other medical conditions and medications; chemotherapeutic agents and antiretrovirals commonly cause neuropathy.
- Family history may identify hereditary neuropathies.
- Social history should include occupation (toxin exposure), sexual history (HIV, hepatitis C), recreational drug use (vasculitis secondary to cocaine), smoking (paraneoplastic disease), and excessive alcohol intake or dietary habits (vitamin deficiencies).
Physical Examination
- Evaluate patterns of sensory loss in specific dermatomal or nerve distributions.
- Diabetic polyneuropathy typically is symmetric in a stocking-glove distribution.
- Large fiber involvement causes vibratory and proprioceptive loss.
- Phalen maneuver (flexing wrists for 60 seconds) or Tinel sign (tapping the median nerve at the wrist) may elicit carpal tunnel syndrome symptoms.53 But both have limited sensitivity and specificity.54
- Diabetic polyneuropathy typically is symmetric in a stocking-glove distribution.
- Evaluate for patterns of weakness and atrophy.
- Toe extension and ankle dorsiflexion weakness may be seen in polyneuropathy.
- Isolated weakness is typical of mononeuropathies and if more generalized may suggest a myopathy.
- If the symptoms of weakness and/or numbness are primarily in the hands, cervical myelopathy should be excluded.
- Toe extension and ankle dorsiflexion weakness may be seen in polyneuropathy.
- Deep tendon reflexes are typically depressed or lost in large fiber neuropathy (ankle jerk lost first), and is more prominent in demyelinating neuropathies.
- Evaluate orthostatic blood pressure to assess for autonomic involvement.
- Gait may be affected in severe neuropathy.
- Signs of upper motor neuron disease are typically absent; if present, they may represent amyotrophic lateral sclerosis or spinal cord disease.
Differential Diagnosis
Many conditions can affect the peripheral nerves (see Table 43-9), and about 25% of cases of chronic polyneuropathy remain unknown. The most common presentations are described below.
TABLE 43-9 Causes of Peripheral Neuropathy
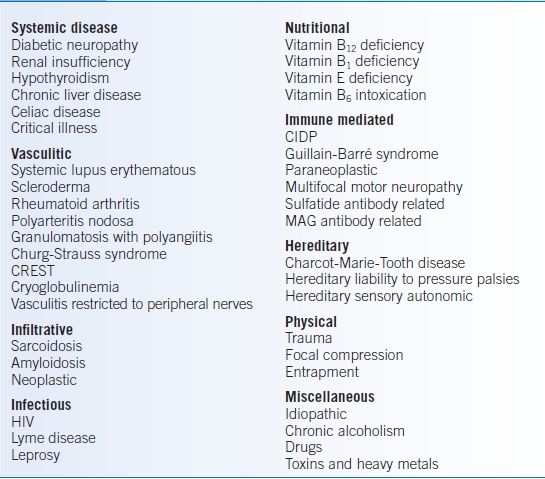
CIDP, chronic inflammatory demyelinating polyneuropathy; CREST, calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia; MAG, myelin-associated glycoprotein.
Diabetic Neuropathy
- Over 30% of diabetic patients have a length-dependent peripheral neuropathy.
- Burning and pain are common symptoms suggesting a small fiber neuropathy, which may progress over months to years.
- Electrodiagnostic studies may be normal early or demonstrate an axonal sensory-motor polyneuropathy that may have autonomic involvement.
- Skin biopsies may show a decrease in nerve fibers distally.
Entrapment Neuropathies
- Systemic diseases like thyroid disease, diabetes, or amyloidosis increase susceptibility to entrapment neuropathies.
- Carpal tunnel syndrome (CTS) is caused by increased pressure within the carpal tunnel producing median nerve ischemia.53
- There is sensation loss in thumb, second, and third digits, lateral half of fourth digit, and the adjacent palmar surface.
- Thenar muscle atrophy can eventually occur.
- Median nerve conduction delay is evident across the wrist on nerve conduction studies (NCS).
- There is sensation loss in thumb, second, and third digits, lateral half of fourth digit, and the adjacent palmar surface.
- The ulnar nerve may become compressed in the retrocondylar groove near the elbow, which becomes more exposed with a flexed elbow and commonly occurs during sleep. Sensory loss occurs in the medial half of the fourth digit, entire fifth digit, and the adjacent palmar surface. Weakness is seen in the first dorsal interosseous muscle.
- Radial nerve entrapment in the radial groove of the forearm results in wrist drop.
- Meralgia paresthetica produces lateral thigh sensory complaints as a result of lateral femoral cutaneous compression at the pelvic brim by obesity and tight-fitting garments.
Trigeminal Neuralgia
- Classic trigeminal neuralgia is generally thought to be due to trigeminal nerve root compression by an overlying blood vessel resulting in demyelination of sensory fibers.
- When bilateral, consider other etiologies such as multiple sclerosis.
- Patients suffer from lancinating pain in distribution of the fifth cranial nerve (third, second, and first divisions in order of most commonly affected).
Bell Palsy
- Bell palsy is a unilateral peripheral facial nerve cranial mononeuropathy causing upper and lower facial weakness with possible alteration of taste and hearing (hyperacusis).
- It is often idiopathic, but etiologies such as Lyme disease, sarcoid, and syphilis should be considered, especially if recurrent or bilateral.
Vasculitic Neuropathy
- Vasculitis neuropathy is a subacute progressive painful sensorimotor axonal neuropathy.
- It classically presents as mononeuritis multiplex but may present as symmetric polyneuropathy.
- It is usually associated with systemic vasculitic disorders but may be confined to the peripheral nerves, causing peripheral nerve infarction secondary to vascular occlusion by inflammation and fibrinoid necrosis.55
- Nerve biopsy may show epineural inflammation, vessel wall pathology, and axonal loss.
Guillain-Barré Syndrome
- Guillain-Barré syndrome (GBS) presents as a rapidly progressive ascending weakness with areflexia and variable sensory involvement. Symptoms peak within 4 weeks.56
- Campylobacter jejuni or cytomegalovirus (CMV) infection may precede the neuropathy.
- Respiratory muscles are often involved; 25% of patients require ventilator support.
- Electrodiagnostic studies may not reveal the typical demyelination for 2 weeks.
- Increased CSF protein is present in approximately 80% of patients.
Chronic Inflammatory Demyelinating Polyneuropathy
- Chronic inflammatory demyelinating polyneuropathy (CIDP) is the most common type of acquired chronic demyelinating polyneuropathy.57
- It affects motor nerves more than sensory nerves.
- The course may be relapsing or progressive, evolving over ≥2 months.
- Electrodiagnostic studies demonstrate reduced conduction velocity and conduction block in multiple nerves indicating demyelination.
- CSF protein is typically moderately elevated.
- Nerve biopsy is considered if there is a high clinical suspicion for CIDP, as electrodiagnostic testing is often nondiagnostic.
HIV Neuropathy
- Distal sensory polyneuropathy is the most common neurologic manifestation of HIV.
- This neuropathy affects >50% of HIV patients.58
- Evidence for axonal sensory neuropathy is demonstrated on electrodiagnostic studies.
- HIV neuropathy remains prevalent despite advent of highly active antiretroviral therapy.
Diagnostic Testing
Laboratories
- Initial laboratory work includes complete blood cell count (CBC), complete metabolic panel (CMP), vitamin B12, folate, TSH, hemoglobin A1C, ESR, and serum protein electrophoresis (SPEP) with immunofixation.
- Additional laboratory work should be considered depending upon the clinical scenario. This includes ANAs, ENAs, rheumatoid factor (RF), urine protein electrophoresis (UPEP), cryoglobulins, quantitative immunoglobulins, HIV, copper, vitamin E, anti-GM1 ganglioside antibodies, anti-sulfatide antibodies, anti–myelin-associated glycoprotein antibodies, and paraneoplastic antibody panel.
Diagnostic Procedures
- Electromyography (EMG) and NCSs are often recommended.
- They can help differentiate axonal versus demyelinating neuropathies.
- The distribution of involvement can be confirmed: mononeuropathy, polyneuropathy, mononeuropathy multiplex, plexopathy, or radiculopathy.
- They can help differentiate axonal versus demyelinating neuropathies.
- If radiculopathy or myelopathy (spinal cord disease) is suspected, MRI of the appropriate level of the spine is recommended.
- Nerve and muscle biopsy may be useful in identifying immune-mediated (e.g., CIDP), inflammatory (e.g., vasculitis), or infiltrative (e.g., amyloidosis) causes of neuropathy.
- Genetic testing is pertinent if a hereditary neuropathy is suspected.
TREATMENT
- If secondary to another disease, aggressive treatment of the underlying etiology (e.g., diabetes, hypothyroidism, vitamin B12 deficiency, and renal insufficiency) may stabilize or improve neuropathic symptoms.
- Physical and occupational therapy can lessen disability. Ankle foot orthotics aid ambulation in patients with foot drop. Weight loss and proper foot care is encouraged, especially if obese and/or diabetic.
- For CTS, wrist splinting, keeping the wrist in a neutral position, is often helpful. Compression often occurs while sleeping and is not limited to overuse. Carpal tunnel release may be indicated with axonal loss or prolonged symptoms.
- Patients with compressive ulnar neuropathy at the elbow should avoid prolonged elbow flexion and leaning on the involved elbow. Nerve transposition may be considered for cases refractory to splinting.
- Treatment of trigeminal neuralgia:
- First-line therapy includes carbamazepine (starting at 200 mg twice daily titrating up to effect). Oxcarbazepine, baclofen, and lamotrigine may also be useful.59
- Microvascular decompression of the nerve should be considered in patients with refractory symptoms.
- First-line therapy includes carbamazepine (starting at 200 mg twice daily titrating up to effect). Oxcarbazepine, baclofen, and lamotrigine may also be useful.59
- Treatment of Bell palsy:
- Corneal protection may be necessary (e.g., Lacri-Lube and an eye patch at night).
- Treatment with oral steroids is beneficial, particularly if started within 3 days of symptom onset.60,61 Consider the addition of acyclovir, valacyclovir, or famciclovir in patients with evidence of herpes infections and severe palsy.62,63
- More than 90% of patients achieve full recovery with good prognostic indicators including incomplete paralysis, early improvement, young age, and preservation of taste. Electrical stimulation of the facial nerve does not hasten the recovery.
- Corneal protection may be necessary (e.g., Lacri-Lube and an eye patch at night).
- Vasculitic neuropathy is treated with corticosteroids or immunosuppression.
- GBS typically requires hospital admission for intravenous immunoglobulins (IVIG) or plasma exchange.56
- CIDP is treated with IVIG, plasma exchange, or corticosteroids.57
- Symptomatic treatment of neuropathic pain64
- Nonopiate agents remain the mainstay of therapy. Tramadol, a partial opioid agonist, and opiates must be prescribed with caution, if needed, given the potential for addiction and abuse.65
- Antiepileptics such as gabapentin, pregabalin, carbamazepine are first-line agents.66–69
- Gabapentin is typically started at 300 mg tid and can be slowly titrated up to 1,200 mg tid (maximum dose 3,600 mg/day). Gabapentin is thought to act via the α2δ subunit of voltage-dependent calcium channels thereby decreasing the release of neurotransmitters. If side effects are a concern gabapentin can be started at 100 mg tid. Dosage and frequency should be decreased in patients with chronic kidney disease. Side effects include fatigue, drowsiness, dizziness, ataxia, tremor, nystagmus, headache, and edema. Data supporting the use of gabapentin specifically for diabetic peripheral neuropathy are conflicting.
- Pregabalin is usually started at 50 mg bid and slowly titrated to a maximum of 150 mg bid, alternatively 100 mg tid. This drug is also thought to act by binding the α2δ subunit. Relatively frequent side effects include fatigue, somnolence, dizziness, confusion, ataxia, headache, tremor, diplopia, and edema.
- Carbamazepine immediate release is started at 100 mg bid and can be increased in 200 mg/day increments to a maximum of 1,200 mg/day. There are FDA boxed warnings for severe skin reactions in patients with the HLA-B*1502 (most often seen in Asians) allele and for agranulocytosis/aplastic anemia. The leukocyte count should be monitored. Adverse reactions also include somnolence, weakness, dizziness, headache, ataxia, rash, and nausea/vomiting.
- Gabapentin is typically started at 300 mg tid and can be slowly titrated up to 1,200 mg tid (maximum dose 3,600 mg/day). Gabapentin is thought to act via the α2δ subunit of voltage-dependent calcium channels thereby decreasing the release of neurotransmitters. If side effects are a concern gabapentin can be started at 100 mg tid. Dosage and frequency should be decreased in patients with chronic kidney disease. Side effects include fatigue, drowsiness, dizziness, ataxia, tremor, nystagmus, headache, and edema. Data supporting the use of gabapentin specifically for diabetic peripheral neuropathy are conflicting.
- Antidepressants such as TCAs (amitriptyline, nortriptyline) and SNRIs (duloxetine, venlafaxine) are other first- or second-line agents.70,71
- Amitriptyline is started at 25 to 50 mg at bedtime and can be titrated up to a maximum of 150 mg/day. Nortriptyline likely has less anticholinergic side effects and is started at 10 to 25 mg at bedtime up to a maximum of 75 mg/day. Desipramine is another reasonable option.
- Duloxetine has an FDA-approved indication for diabetic neuropathy; the starting dose is 30 mg/day, and then increased to 60 mg/day. Higher doses are not additionally effective.
- Venlafaxine, like duloxetine, is an SNRI and is presumed to be effective at a dosage range between 75 and 225 mg/day (if immediate release, in 2 or 3 divided doses).
- SSRIs are not known to useful for neuropathic pain.
- Amitriptyline is started at 25 to 50 mg at bedtime and can be titrated up to a maximum of 150 mg/day. Nortriptyline likely has less anticholinergic side effects and is started at 10 to 25 mg at bedtime up to a maximum of 75 mg/day. Desipramine is another reasonable option.
- Topical lidocaine and capsaicin may provide additional pain control.
- Nonopiate agents remain the mainstay of therapy. Tramadol, a partial opioid agonist, and opiates must be prescribed with caution, if needed, given the potential for addiction and abuse.65
Dystonia
- Dystonias are characterized by involuntary, sustained, patterned, and often repetitive muscle contractions leading to repetitive twisting movements and abnormal postures of the trunk, neck, face, or arms and legs.72
- Dystonias can be triggered by certain actions and relieved by so-called sensory tricks (e.g., touching the side of one’s face), leading to the erroneous impression that the disorder is psychogenic.
- Early-onset (<25 years) forms of dystonia typically begin focally and gradually spread to involve other body regions, becoming progressively debilitating. Adult-onset (>25 years) forms usually begin within craniofacial or cervical musculature and do not migrate or progress significantly over time.
- Common focal dystonias include blepharospasm, task-specific limb dystonias (e.g., writer’s cramp), cervical dystonia (e.g., spasmodic torticollis), vocal cord dystonia (e.g., spasmodic dysphonia), and oromandibular dystonia.72
- Patients with early onset dystonia should be tried on carbidopa/levodopa to evaluate for dopa-responsive dystonia, a rare genetic form of dystonia. Anticholinergics, benzodiazepines, baclofen, dopaminergic agents, dopamine-depleting agents, and tetrabenazine a vesicular monoamine transporter 2 inhibitor) may result in some benefit.
- Botulinum toxin injection to affected muscles is the treatment of choice for focal dystonias.
Essential Tremor
GENERAL PRINCIPLES
- Essential tremor is the most common neurologic cause of postural (e.g., maintaining posture with hands outstretched) or action (e.g., intentionally touching nose with index finger) tremor.
- Severe essential tremor may have a rest component.
- Essential tremor may begin on one side but eventually becomes bilateral/symmetrical.
- Approximately 50% have an autosomal dominant pattern of inheritance.
Clinical Presentation
- Patients may report improvement of essential tremor following alcohol intake.
- Postural or action tremors have a broad differential. Worsening of tremor despite therapeutic trials or development of bradykinesia, rigidity, and postural instability should prompt further evaluation and referral to a movement disorder clinic.
- Patients with Parkinson disease (PD) may have a postural/action tremor.
- Exaggerated physiologic tremor is a commonly observed postural tremor in anxiety, stimulants (e.g., nicotine, caffeine), thyrotoxicosis, and drug or alcohol toxicity or withdrawal.
- Medication-induced tremor is common in the setting of antidepressants, β-agonists, steroids, valproate, lithium, neuroleptics, metoclopramide, theophylline, and levothyroxine.73
- Wilson disease should be considered in any patient under the age of 40 with tremor or other involuntary movements or postures (check serum ceruloplasmin).
- Screening for endocrine and toxic causes should include hyperthyroidism, hypoglycemia, caffeine consumption, nicotine use, chronic alcohol consumption, and medications.
TREATMENT
- Propranolol (immediate or sustained release): titrate to effect (usually 160 to 320 mg).74
- Primidone: begin 62.5 mg daily and titrate to effect, usually 62.5 to 1,000 mg daily.74
- Second-line medications include gabapentin, topiramate, and clonazepam.
- Deep brain stimulation is highly effective for medically refractory cases.
Parkinson Disease and Related Conditions
GENERAL PRINCIPLES
- Parkinsonism is a syndrome characterized by tremor, bradykinesia, rigidity, and postural instability. PD is the most common cause of parkinsonism.
- PD is a common neurodegenerative disease associated with progressive loss of dopaminergic neurons in substantia nigra of the brainstem and presence of Lewy bodies.
- Dementia is a common feature of PD. PD can coexist with other dementias (e.g., Alzheimer disease, vascular dementia).
- If dementia begins early in the course, consider other forms of parkinsonism.
- Typical age of onset is around 60, but there is considerable variability.75
- About 10% of cases are genetic; this is more common in early-onset patients.
- Dementia is a common feature of PD. PD can coexist with other dementias (e.g., Alzheimer disease, vascular dementia).
- Secondary parkinsonism may result from agents with dopamine-blocking properties (e.g., antipsychotics and antiemetics).
- Ninety percent begin within 3 months of starting the offending agent.76
- Offending agents should be withdrawn gradually to avoid reemergent dyskinesia.
- Ninety percent begin within 3 months of starting the offending agent.76
- Other neurodegenerative forms of parkinsonism (so-called Parkinson-plus syndromes) are rarer and less likely to respond to dopaminergic drugs.77
- These include dementia with Lewy bodies, progressive supranuclear palsy, multiple system atrophy, and corticobasal degeneration.
- Early and severe postural instability and/or dementia are clues for these syndromes; there is extensive clinical overlap between these and PD.
- These include dementia with Lewy bodies, progressive supranuclear palsy, multiple system atrophy, and corticobasal degeneration.
- Normal pressure hydrocephalus may also mimic parkinsonism. The clinical triad consists of dementia, urinary incontinence, and a magnetic gait.
DIAGNOSIS
- History and examination should focus on clinical features of parkinsonism.
- Tremor occurs predominantly at rest.
- Bradykinesia.
- Rigidity is often described as lead pipe.
- Postural instability can contribute to falls.
- Tremor occurs predominantly at rest.
- Symptoms in PD often begin on one side and remain asymmetric until later in the disease. Consider other etiologies of parkinsonism if clinical features are symmetric.
- Other common motor findings include shuffling gait, stooped posture, loss of facial expression, decreased blinking, decreased arm swing, and occasional freezing of gait.
- Common nonmotor symptoms include cognitive dysfunction/dementia, psychosis and hallucinations, mood disorders, depression, anxiety, apathy and abulia, sleep disorders, excessive daytime somnolence, fatigue, autonomic dysfunction, olfactory dysfunction, sensory complaints, and seborrhea.78
TREATMENT
- Carbidopa/levodopa is the most effective symptomatic therapy.79,80
- Other dopaminergic agents such as MAOI and catechol-O-methyl transferase (COMT) inhibitors are also used.81
- Early treatment of PD with levodopa does not accelerate the course of the disease and may even slow it down.82
- Other dopaminergic agents such as MAOI and catechol-O-methyl transferase (COMT) inhibitors are also used.81
- Dopamine agonists (e.g., pramipexole, ropinirole) may be associated with less dyskinesia but may increase risk of peripheral edema, somnolence/sudden sleep, constipation, hallucinations, nausea, and impulse control disorders.83
- PD is progressive, requiring gradual increases in medication over time.
- Treatment complications include dyskinesias and motor fluctuations. Other common side effects include somnolence, orthostatic hypotension, hallucinations, psychosis, anorexia, and nausea.
- Dopamine antagonists (antiemetics, antipsychotics, etc.) and anticholinergics can worsen parkinsonism.
- Ondansetron should be used for nausea. Domperidone, which is not available in the US, can be used in refractory cases.
- Low-dose quetiapine (starting 12.5 to 25 mg at night) should be reserved for refractory psychosis.
- Ondansetron should be used for nausea. Domperidone, which is not available in the US, can be used in refractory cases.
- Medically refractory cases of PD may be treated with deep brain stimulation.84
Dementia
GENERAL PRINCIPLES
- 10% of persons over age 65 and up to 50% over 85 have dementia.
- Dementia is a deterioration in cognitive, reasoning, and language abilities of sufficient severity to interfere with activities of daily living.
- Mild cognitive impairment (MCI) is mild memory loss that may be evident on standardized testing but causes minimal impairment of activities of daily living. Between 6% and 25% of patients with MCI progress to dementia each year.
- Delirium is an acute impairment in awareness and orientation with disturbances of perception (i.e., hallucinations) in which symptoms typically wax and wane. In contrast, dementia has a more chronic steady decline in memory with clear sensorium.
- Dementia commonly results from a primary degenerative process resulting in accumulation of abnormal proteins in the brain. Alzheimer disease is the most common form.
DIAGNOSIS
Clinical Presentation
History
- The newly released Diagnostic and Statistical Manual (DSM) V now defines criteria for major and mild neurocognitive disorders.85
- Major neurocognitive disorder must meet the following criteria85:
- Evidence of a significant cognitive decline from prior performance in at least one cognitive domain (learning/memory, language, complex attention, executive function, and social cognition). Ideally this should be confirmed with neuropsychologic testing or some other quantified clinical assessment.
- Cognitive deficits interfere with typical activities (at least requiring assistance with executive functioning tasks).
- Cognitive deficits are not exclusively due to delirium.
- Cognitive deficits not better explained by another mental disorder.
- Evidence of a significant cognitive decline from prior performance in at least one cognitive domain (learning/memory, language, complex attention, executive function, and social cognition). Ideally this should be confirmed with neuropsychologic testing or some other quantified clinical assessment.
- Mild neurocognitive disorder must meet these criteria85:
- Evidence of modest cognitive decline from prior performance (learning/memory, language, complex attention, executive function, and social cognition).
- Cognitive deficits do not interfere with independent activities of daily living.
- Cognitive deficits are not exclusively due to delirium.
- Cognitive deficits not better explained by another mental disorder.
- Evidence of modest cognitive decline from prior performance (learning/memory, language, complex attention, executive function, and social cognition).
- Interview both the patient and a collateral source.
- Review prescription and nonprescription medications as possible etiologies.
Physical Examination
- Cognitive screening instruments (short blessed, AD8, mini mental status examination, or Montreal Cognitive Assessment) are easy to administer and follow over time.86,87
- Cardinal features of Alzheimer disease on mental status examination include amnestic memory loss, language deterioration, and visual-spatial impairment.88 Motor, sensory, and gait deficits can be seen in later stages.
- Evaluate the signs of parkinsonism (resting tremor, rigidity, bradykinesia, postural instability), which may suggest a Parkinson-plus syndrome (see “Parkinson Disease” section).
- Primitive reflexes called frontal release signs may reemerge (e.g., palmomental, glabellar, grasp, snout, and suck).
Differential Diagnosis
- Table 43-10 presents the differential diagnosis of dementia.
- Alzheimer disease is the most common progressive neurodegenerative disorder with prominent memory, cognitive, and visual-spatial deficits.
- Dementia related to parkinsonism has a clinical and pathologic overlap with Alzheimer disease and PD. In addition to memory and cognitive difficulties, patients exhibit signs of parkinsonism with rigidity, bradykinesia, postural instability, and tremor. Visual hallucinations may be prevalent.
- Frontotemporal dementia is commonly misdiagnosed as Alzheimer disease. It tends to present with prominent behavioral (e.g., disinhibition, impulsivity, apathy, and loss of insight) or language abnormalities (e.g., nonfluent or fluent aphasias).89
- Vascular dementia classically involves a step-like progression of symptoms with evidence of multiple strokes on brain imaging.
- Creutzfeldt-Jakob disease is a rapidly progressive dementing disease often accompanied by myoclonus. It is a prion disease with familial and sporadic forms. Definitive diagnosis can only be made by brain biopsy or at autopsy, but LP, MRI (diffusion imaging), and EEG findings can be helpful.90
TABLE 43-10 Causes of Dementia

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


