Fig. 35.1.
Renal cell carcinoma, clear cell type. Tumor is typically well demarcated from normal kidney by a fibrous pseudocapsule, and the cut surface is usually variegated. Viable tumor is golden yellow, in a background of cystic and myxoid degeneration, hemorrhage, and necrosis.
Microscopic
♦
Typically composed of cells with abundant cytoplasm that appears optically clear (due to loss of intracellular glycogen and lipids during histologic processing) and with distinct cell membranes
♦
Tumor architecture is diverse – solid, alveolar, and acinar patterns are often admixed
♦
Tumor cells are set in a regular network of small delicate blood vessels (Fig. 35.2)
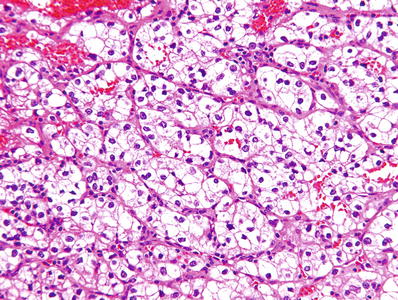
Fig. 35.2.
Renal cell carcinoma, clear cell type. Classic clear cell carcinoma is composed of cells with distinct cell membranes and optically clear cytoplasm. Tumor cells are arranged in sheets, compact nests, alveolar, acinar, and microcystic or even macrocystic structures, separated by an abundance of thin-walled blood vessels.
♦
Microcysts and macrocysts containing eosinophilic fluid may be present (Fig. 35.3)
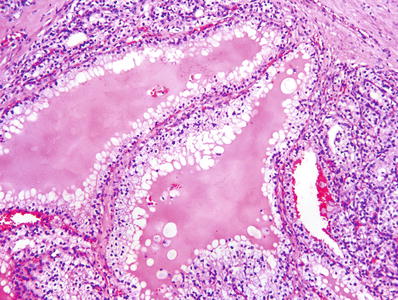
Fig. 35.3.
Renal cell carcinoma, clear cell type. Tubular structures vary in size. Microcysts contain eosinophilic fluid or extravasated red blood cells.
♦
Many cytologic variations may be seen
Cells with granular eosinophilic cytoplasm
Cells with rhabdoid morphology
Interlacing or whorled bundles of spindle cells, sometimes with marked nuclear pleomorphism (sarcomatoid change)
♦
ISUP nuclear grade is based on nucleolar prominence for grades 1–3 tumors; grade 4 tumors are defined by the presence of parameters listed below
Grade 1: nucleoli absent or inconspicuous at 400× (Fig. 35.4)
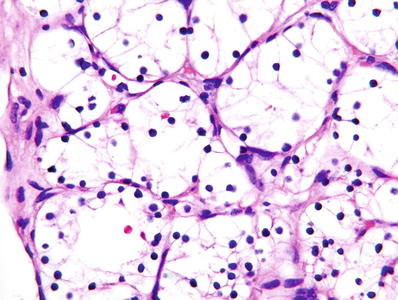
Fig. 35.4.
Clear cell renal cell carcinoma, ISUP grade 1. At 400× magnification, nucleoli are absent or inconspicuous.
Grade 2: nucleoli distinctly visible at 400× but inconspicuous or invisible at 100× (Fig. 35.5)
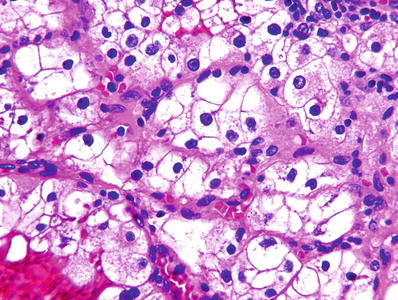
Fig. 35.5.
Clear cell renal cell carcinoma, ISUP grade 2. Nucleoli are inconspicuous or invisible at 100× but are distinctly visible at 400×, as shown here.
Grade 3: nucleoli are conspicuous at 100× (Fig. 35.6)
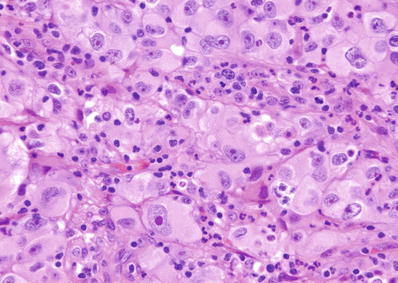
Fig. 35.6.
Clear cell renal cell carcinoma, ISUP grade 3. The large prominent nucleoli shown in this image (at 400×) were easily identified at 100× magnification, compatible with ISUP grade 3.
Grade 4: pronounced nuclear pleomorphism and/or tumor giant cells (Fig. 35.7) and/or rhabdoid morphology and/or sarcomatoid morphology (Fig. 35.8)
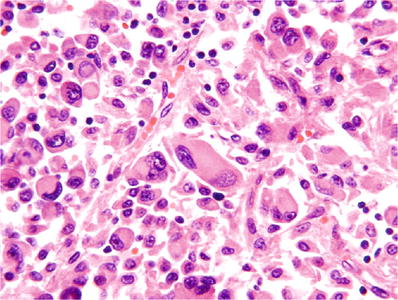
Fig. 35.7
Clear cell renal cell carcinoma, ISUP grade 4. Tumor cells with extreme pleomorphism are present.
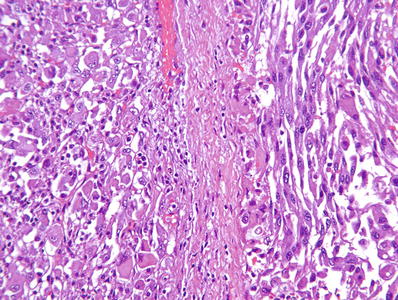
Fig. 35.8.
Clear cell renal cell carcinoma , ISUP grade 4. Tumor cells show rhabdoid (left side of image) or sarcomatoid (right side of image) morphology.
♦
Incidence of immunoreactivity to antibodies against the following antibodies is as indicated:
PAX8 (95–100%)
Carbonic anhydrase IX (CA IX), 75–100%
Frequently positive: keratins AE1/AE3 and CAM5.2, vimentin, RCC marker, CD10, and EMA
♦
Rarely expressed: CK7 and high molecular weight cytokeratins (14, 34βE12)
Table 35.1.
Immunohistochemical Findings in Renal Epithelial Neoplasms
CD10 | CA IX | RCC marker | Vimentin | EMA | CK7 | CK20 | HMWCK | CD117 (c–kit) | AMACR | |
|---|---|---|---|---|---|---|---|---|---|---|
Clear cell renal cell carcinoma | +(81–100%) | + | +(85%) | + | +/− (20–90%) | – | − | −/+f | −/+f | −/+f |
Papillary renal cell carcinoma | +(63–93%) | +/−f | +(93%) | +/− | +/− (30–45%) | + | +/− | −/+f | −/+f | + |
Clear cell papillary renal cell carcinoma | − | +(cup-like) | +/− | + | +/− | + | − | − | − | – |
Chromophobe renal cell carcinoma | −/+f | − | +(50%) | – | + | + | − | − | + | − |
Oncocytoma | −/+f | − | – | – | + | Focal (5% or less)a | +/− | − | + | −/+f |
Collecting duct carcinoma | – | +/−f | – | + | +/− | +/− | − | + | − | − |
Urothelial carcinoma | −/+f | −/+ | – | – | + | + | + | + | −/+f | −/+f |
Multilocular Cystic Renal Neoplasm of Low Malignant Potential
Clinical
♦
Occurs in adults, with a mean age of 51 years (range, 20–76)
♦
90% or more are discovered incidentally
♦
Accounts for less than 1% of all renal tumors
♦
Male to female ratio is about 2:1
♦
No recorded instances of recurrence or metastasis after excision
Macroscopic
♦
Well circumscribed, with fibrous capsule
♦
Composed entirely of cysts containing clear, serous, gelatinous, or hemorrhagic fluid
♦
Cysts are separated by irregular septa of variable thickness
♦
Grossly evident solid nodules of tumor exclude the diagnosis
Microscopic
♦
The cells lining the cysts form a single layer, although, occasionally, small papillae may extend into the cysts (Fig. 35.9)
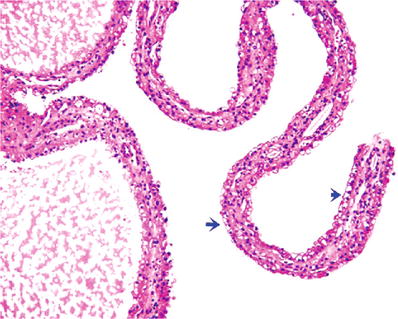
Fig. 35.9.
Multilocular cystic neoplasm of low malignant potential. Delicate septa between the cystic spaces are lined by clear cells (blue arrows) and flat cuboidal cells.
♦
Tumor cells have plentiful clear cytoplasm and small nuclei that lack nucleoli (ISUP grade 1)
♦
Septa consist of fibrous tissue, sometimes with calcific or osteoid deposits
♦
Septa contain nonexpansile aggregates of clear cells similar to those that line the cysts (Fig. 35.10)
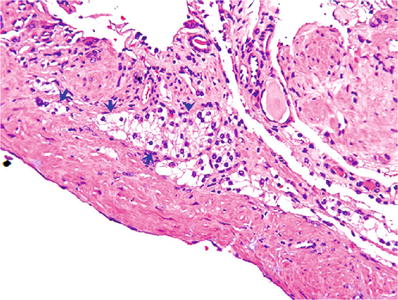
Fig. 35.10.
Multilocular cystic neoplasm of low malignant potential. Aggregates of clear cells are present in the septa; the aggregates are noted only by microscopic examination; they are not “expansile”.
♦
Sarcomatous change, vascular invasion, and necrosis exclude the diagnosis
Genetics
♦
Probably genetically related to clear cell renal cell carcinoma
♦
74% has chromosome 3p deletions, similar to those identified in 89% of clear cell renal cell carcinoma
♦
VHL mutations have been found in 25% of cases
Papillary Renal Cell Carcinoma
Clinical
♦
Male to female ratio is about 2:1; affects patients from pediatric age to great old age; mean patient age is 59–63 years
♦
Accounts for up to 18.5% of all RCCs; it is the second most common type of renal cell carcinoma
♦
Using the 2016 WHO criteria, papillary or tubulopapillary tumors greater than 1.5 cm in diameter are considered carcinoma
♦
Unencapsulated tumors with papillary or tubular architecture of low nuclear grade (grades 1–2) and ≤1.5 cm in size are considered benign (papillary adenoma; see further discussion in the “Papillary Adenoma” section)
♦
Hereditary papillary RCC is rare and is related to a germline mutation of the c–met gene
♦
Cytogenetics
Trisomy or tetrasomy 7, trisomy 17, loss of chromosome Y most commonly
Trisomy of chromosomes 8, 12, 16, and 20 common
Wide variety of other chromosomal aberrations also reported
♦
Prognosis
Overall, better than for carcinomas of clear cell, collecting duct, or HLRCC type
Prognosis for type 1 tumors is better than for type 2 tumors
Prognosis is worsened by the presence of sarcomatoid or rhabdoid morphology in the tumor
Macroscopic
♦
Well-circumscribed cortical tumor, often with a thick fibrous pseudocapsule
♦
Solid tumors often have a friable consistency (Fig. 35.11)
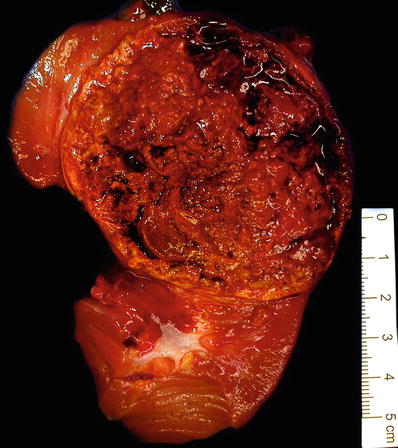
Fig. 35.11.
Papillary renal cell carcinoma . A thick fibrous pseudocapsule is present in up to two-thirds of cases. Many such tumors are extensively hemorrhagic and friable in consistency. Infrequently the thick pseudocapsule encloses little more than old bloody necrotic material, and extensive tumor sampling is needed to identify viable tumor.
♦
Hemorrhage, necrosis, and cystic degeneration are common
♦
More often multiple than other types of RCC
♦
May contain golden yellow areas reflecting macrophage content
Microscopic
♦
Tumor is composed of papillary structures and tubules in varying proportions
♦
Papillary cores are lined by a single cell layer that may show pseudostratification
♦
Fibrovascular cores may be sclerotic, edematous, or expanded by foamy or hemosiderin-laden macrophages, psammoma bodies, or cholesterol clefts
♦
Two morphologic variants
Type 1: characterized by a single layer of small dark cells with scant cytoplasm and low nuclear grade (Fig. 35.12)
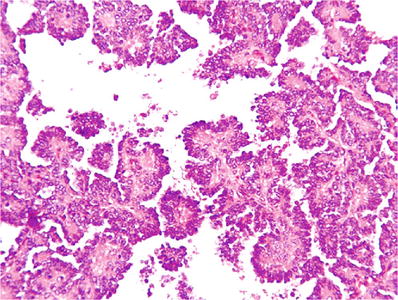
Fig. 35.12.
Papillary renal cell carcinoma, type 1. Tumor is composed of papillary structures lined by small dark uniform cells with minimal cytoplasm and grade 1 nuclear characteristics.
Type 2: cells lining the papillae are large and pseudostratified, with more abundant pink cytoplasm, and of higher nuclear grade (Fig. 35.13)
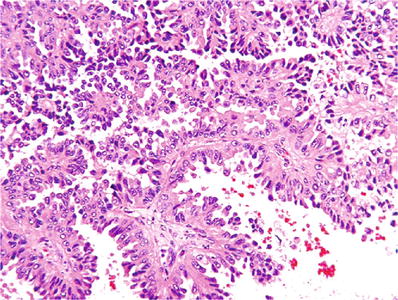
Fig. 35.13.
Papillary renal cell carcinoma, type 2. Tumor is composed of papillary structures lined by pseudostratified cells with abundant pink cytoplasm and high-grade (grade 3) nuclear characteristics.
♦
Tubules may be compacted to form “parallel arrays” (Fig. 35.14)
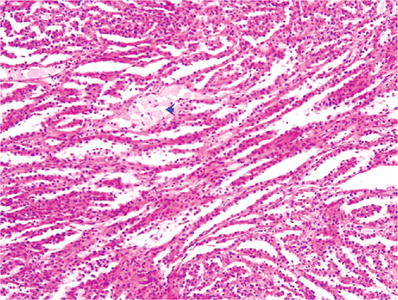
Fig. 35.14.
Papillary renal cell carcinoma. The papillary structures are compressed and form “parallel arrays.” Arrow indicates stromal macrophages.
♦
Papillary stalks often contain abundant foamy macrophages (Fig. 35.15)
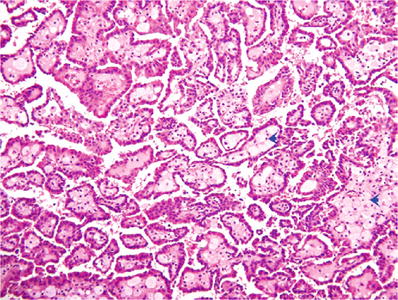
Fig. 35.15.
Papillary renal cell carcinoma . In this example, numerous foamy macrophages are present in the fibrovascular cores, a feature that is characteristic of this type of RCC.
♦
Sarcomatoid change may be observed in up to 5% of cases
♦
The ISUP grading system is applicable to papillary renal cell carcinoma
Immunohistochemistry
♦
Tumor cells are frequently immunoreactive to antibodies against pancytokeratins, CK7, AMACR, vimentin, CD10, and RCC antigen
Hereditary Leiomyomatosis Renal Cell Carcinoma (HLRCC)-associated Renal Cell Carcinoma
Clinical
♦
Rare and aggressive tumor
♦
Arises in patients with an autosomal dominant tumor syndrome associated with germline mutations in the fumarate hydratase (FH) gene at chromosome 1q42
♦
Syndrome is known as the hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome
♦
Patients typically develop painful cutaneous leiomyomas, and females develop uterine leiomyomas, often at an unusually young age, and, rarely, uterine leiomyosarcomas
♦
Diagnosis is confirmed by detection of germline mutations in the gene encoding fumarate hydratase (FH, 1q42.3-q43)
♦
HLRCC syndrome has a lower penetrance of RCC; about 33% of patients with HLRCC syndrome develop renal cell carcinoma
♦
Those who develop renal cell carcinoma typically present with advanced-stage disease, and the majority die of renal cancer
♦
If the tumor is recognized, counseling and follow-up of family members can be initiated and may be greatly beneficial to them – identification and resection of even small tumors may be lifesaving
Macroscopic
♦
With rare exceptions, tumors are unilateral and solitary; ranging in size from 2.3 cm to 20 cm
♦
Tumors may be predominantly solid, predominantly cystic, or have a combination of solid–cystic components
Microscopic
♦
Tumors typically exhibit papillary or tubulopapillary architecture
♦
Morphology may closely mimic that of type 2 papillary renal cell carcinoma
♦
Tumor cells have abundant eosinophilic cytoplasm and large nuclei with a very prominent inclusion-like orangiophilic or eosinophilic nucleoli, surrounded by a clear halo (Fig. 35.16)
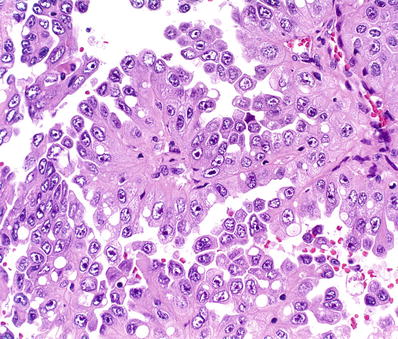
Fig. 35.16.
Hereditary leiomyomatosis renal cell carcinoma. Tumor morphology is similar to that of conventional type 2 papillary renal cell carcinoma, but many of the large nuclei exhibit very prominent inclusion-like orangiophilic or eosinophilic nucleoli, surrounded by a clear halo, a finding that should raise concern for this aggressive type of renal cancer.
Other studies
♦
Immunohistochemistr y: loss of FH staining and overexpression of modified cysteine (2SC)
♦
Somatic mutations in FH are rare in sporadic type 2 papillary renal cell carcinoma
Chromophobe Renal Cell Carcinoma
Clinical
♦
Accounts for about 5–7% of all resected renal cell carcinomas
♦
Believed to originate from intercalated cells of collecting duct epithelium
♦
Males and females are almost equally affected; mean patient age is about 55 years
♦
Cytogenetics: Typically, extensive loss of chromosomes (−1, −2, −6, −10, −13, −17, −21) with hypodiploid DNA index
♦
Commonly of low stage when detected
♦
Outcome predicated by tumor stage and the presence or absence of sarcomatoid transformation, tumor necrosis, and small vessel invasion
♦
Overall prognosis is relatively favorable: reported 5 year survival ranges from 78% to 100%
Macroscopic
♦
Typically solid, light tan or mahogany brown, and well circumscribed (Fig. 35.17)
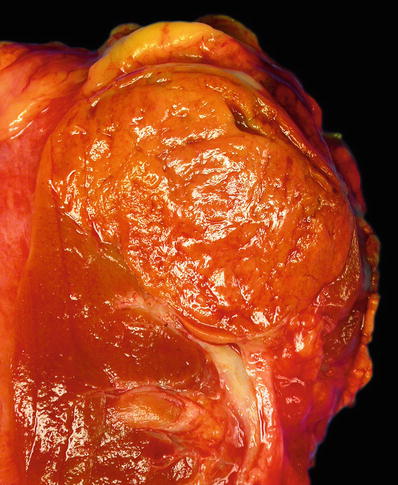
Fig. 35.17.
Chromophobe renal cell carcinoma. A solitary well-circumscribed tumor with a solid, homogeneous, and light brown cut surface which is shown here. Less often the tumor is gray tan, light pink, or yellow white.
♦
Necrosis, hemorrhage, and scarring are minimal or absent
Microscopic
♦
Growth pattern is generally solid but sometimes limited tubule formation is evident
♦
Sheets of cells are intersected by fibrovascular septa, which may be thin and delicate but are often broad and hyalinized
♦
Tumors are composed of varying proportions of two cell types
“Classic cell type” (Fig. 35.18):
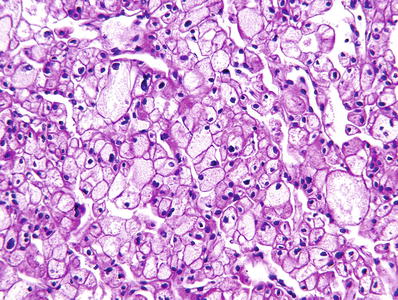
Fig. 35.18.
Chromophobe renal cell carcinoma, “classic cell type”: large polygonal cells with thick distinct “plant-like” cell membranes and abundant pale or translucent flocculent pink cytoplasm.
Large polygonal cells, with thick distinct “plant-like” cell membranes
Tumor cells have abundant pale or translucent flocculent pink cytoplasm
The largest tumor cells tend to aggregate adjacent to fibrovascular septa
“Eosinophilic cell type” (Fig. 35.19):
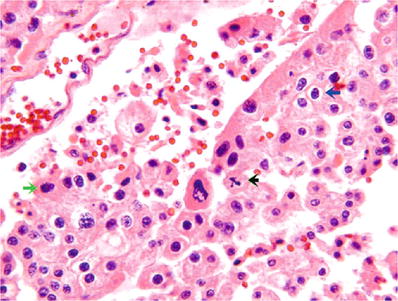
Fig. 35.19.
Chromophobe renal cell carcinoma, “ eosinophilic cell type.” Cytoplasm is less abundant and is darkly eosinophilic. Blue arrow indicates a round regular nucleus with characteristic perinuclear halo; black arrow indicates a mitotic figure; green arrow indicates an irregular “raisinoid” nucleus.
Smaller cells, arranged in small nests, or intermingled with “classic” cells
Cytoplasm is less abundant and is granular and more darkly eosinophilic
Distinctive perinuclear halos are often present due to the accumulation of cytoplasmic organelles around nuclei
♦
In either cell type, nuclei may be round and uniform or irregular and “raisinoid”
♦
ISUP grading is not valid for this type of renal cell carcinoma
♦
Sarcomatoid transformation is observed in 2–8% cases; the presence of this finding markedly worsens the prognosis
Histochemical and Immunohistochemical Studies
♦
Hale colloidal iron stain for acid mucopolysaccharides variable but strongly and diffusely positive in many cases; technically a difficult stain to perform, interpretation may be difficult as well
♦
Typically positive immunostaining for CK7, CD117, parvalbumin, and kidney-specific cadherin
♦
Usually negative immunostaining for vimentin
Ultrastructure
♦
Numerous cytoplasmic microvesicles (150–300 nm) of unknown origin
Collecting Duct Carcinoma
Clinical
♦
Accounts for less than 1–2% of renal cancers
♦
Male to female ratio is about 2:1; mean age is about 55 years (range, 13–85 years)
♦
Nearly 33% of patients have metastases at the time of diagnosis and more than 70% are at pathologic stage pT3 or higher when diagnosed
♦
Cytogenetics: loss of heterozygosity on multiple chromosome arms (1q, 3p, 6p, 8p, 13q, 21q) and losses of entire chromosomes are but a few of a wide variety of reported abnormalities
♦
Prognosis: Poor; about 67% of patients die within 2 years after diagnosis
Macroscopic
♦
Tumors are typically gray white and range in size from 2.5 cm to 15 cm, with an average of 5 cm
♦
When small, they are centered in the renal medulla
♦
Tumors exhibit irregular and grossly infiltrative borders
Microscopic
♦
Composed of tubular or tubulopapillary structures
♦
Lined by cytologic malignant cells, some may show a hobnail appearance
♦
Irregular angulated glands infiltrate renal parenchyma
♦
Infiltration is accompanied by prominent stromal desmoplasia and frequently by a pronounced chronic inflammatory response at the interface between advancing tumor and normal adjacent renal parenchyma (Figs. 35.20 and 35.21)
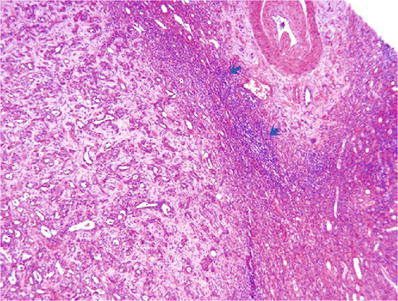
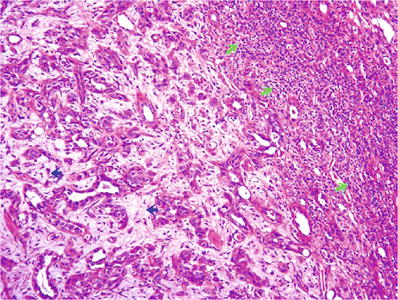
Fig. 35.20.
Collecting duct carcinoma . This low power view illustrates the tubular architecture of the tumor, stromal desmoplasia, and the “pushing” border, with pronounced inflammation (blue arrows) at the interface between tumor and normal kidney.
Fig. 35.21.
Collecting duct carcinoma . Tumor is composed of irregular tubules in a desmoplastic stroma (blue arrows), with considerable peritumoral chronic inflammatory cell infiltrate (green arrows).
♦
Intraluminal and intracytoplasmic mucin may be seen
♦
Dysplasia may be noted in the epithelium of adjacent collecting ducts (Fig. 35.22)
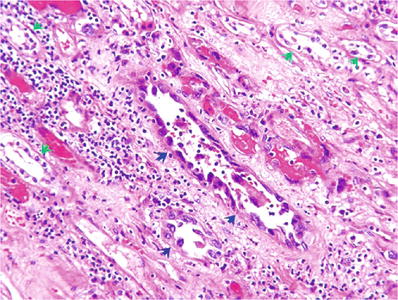
Fig. 35.22.
Collecting duct carcinoma , with high-grade nuclear dysplasia in some native renal tubules (blue arrows); normal tubules are indicated by green arrows.
♦
Sarcomatoid change is common, occurring in up to 30% of cases
♦
Difficult to separate from urothelial carcinoma, adenocarcinoma of renal pelvis, metastatic adenocarcinoma, renal medullary carcinoma, and type 2 papillary renal cell carcinoma
Immunohistochemistry
♦
Typically expresses high molecular weight keratins: CK7, CK19, and 34βE12
♦
Recommended panel to differentiate from mimics: PAX2, PAX8, INI1, OCT3/4, and p63
Renal Medullary Carcinoma
Clinical
♦
Most often occurs in young persons with sickle cell trait, sickle cell disease, or hemoglobin sickle cell disease
♦
Male to female ratio is about 2:1
♦
The great majority of patients (90%) are African-American, Hispanic, or Brazilian; only 5% is Caucasian
♦
Age range is 5–69 years; most are in second and third decades of life
♦
Weight loss, hematuria, flank or abdominal pain, or palpable mass are common symptoms and signs
♦
Often, the presenting symptoms are related to metastases
♦
Tumor is believed to originate in collecting duct epithelium
♦
Genetics: increased HIF1A expression, deregulated DNA remodeling and repair, LOH or deletions at SMARCB1, and some similarities to urothelial carcinoma
♦
Prognosis is poor: most patients die within 1 year of diagnosis, and mean survival is only about 15 weeks
Macroscopic
♦
Gray white, grossly infiltrative, and typically centered in the renal medulla, 4–12 cm (mean, 7 cm), with a predilection for right kidney
Microscopic
♦
A variety of architectural patterns may be present
Reticular or yolk-sac appearance
Adenoid cystic appearance
Solid sheets, nests, or tubules
♦
Tumor cells may show rhabdoid morphology
♦
A strikingly prominent infiltrate of neutrophils may be evident within the tumor, and mucin production by tumor cells is often observed (Fig. 35.23)
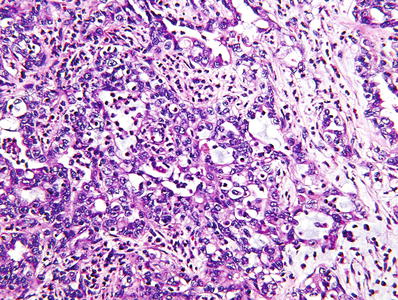
Fig. 35.23.
Renal medullary carcinoma . This type of RCC commonly demonstrates several different morphologic patterns. The malignant tubules in this example are similar to those seen in collecting duct carcinoma.
♦
Sickled erythrocytes are evident in the great majority of cases (Fig. 35.24)
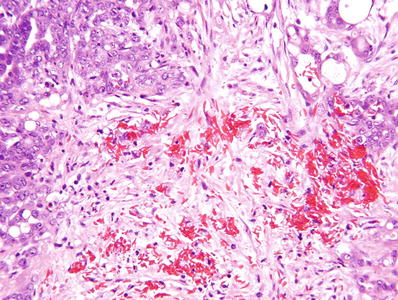
Fig. 35.24.
Renal medullary carcinoma . Arrows indicate sickled red cells in a background of poorly differentiated renal carcinoma.
Immunohistochemistry
♦
Tumor cells usually show positive immunostaining for PAX8, CK7, polyclonal CEA, keratin CAM5.2, and frequently OCT3/4
♦
INI1 immunostain is negative in tumor cells due to deletions at SMARCB1 locus
MIT Family Translocation Renal Cell Carcinoma
♦
TFE3 and TFEB are members of the MIT subfamily of transcription factors
♦
Gene fusions involving TFE3 are associated with Xp11 translocation carcinomas of the kidney
♦
A MALAT1–TFEB gene fusion is associated with t(6;11) translocation carcinomas of the kidney
Xp11 Translocation Renal Cell Carcinoma
♦
Accounts for about 40% of pediatric renal cell carcinomas and up to 4% of adult renal carcinomas
♦
Tumors are defined by a variety of chromosome translocations , all of which involve a breakpoint at Xp11.2 and all of which result in the fusion of any one of multiple translocation partners with the TFE3 transcription factor gene at this locus
t(X;1)(p11.2;q21) translocation fuses the PRCC and TFE3 genes
t(X;1)(p11.2;p34) fuses the PSF and TFE3 genes
inv(X)(p11;q12) fuses the NonO (p54 nrb ) and TFE3 genes
t(X;17)(p11.2;q23) fuses the CLTC and TFE3 genes
♦
A balanced t(X;17)(p11.2;q25) in one variant of translocation carcinoma; it shares the same ASPSCR1–TFE3 gene fusion as alveolar soft part sarcoma – der(17)t(X;17)(p11.2;q25), which fuses the TFE3 gene to a novel gene, ASPL, on 17q25
Clinical
♦
Majority of patients are less than 50 years old, but patients up to 79 years old have been reported
♦
Equally frequent in males and females
♦
Some patients have a history of prior chemotherapy treatment
♦
Half of patients present with abdominal or flank pain or hematuria; some tumors are detected as a palpable mass, and some are found incidentally
♦
Prognosis: Similar to that of patients with clear cell RCC but worse than for those with papillary RCC
♦
Outcome is worse in older patients and in those with metastatic disease at presentation
Morphology
♦
Circumscribed 3–14 cm and may or may not be encapsulated
♦
Typically, papillary and nested architecture; some have abundant psammoma bodies (Fig. 35.25)
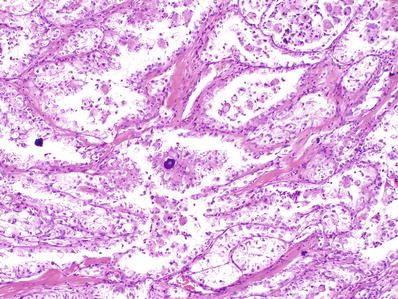
Fig. 35.25.
Xp11 translocation renal cell carcinoma (ASPSCR1–TFE3 type). Tumor shows nested to pseudopapillary architecture and numerous psammomatous calcifications . Tumor cells possess voluminous clear cytoplasm. When examined at higher power, they have vesicular nuclei with prominent nucleoli.
♦
Tumor cells are epithelioid, with sharply defined cell borders and with abundant cytoplasm that varies from clear and finely granular to distinctly eosinophilic
♦
Areas of necrosis are seen in the majority of cases, but mitotic activity is very limited
♦
In rare cases, melanin pigment may be present
Immunohistochemistry and FISH
♦
Cytokeratins and EMA are underexpressed
♦
PAX8 consistently positive
♦
Cathepsin K positive in 60%; melanocytic markers positive rarely
♦
Strong nuclear immunostaining for TFE3 is highly sensitive and specific but technically difficult in formalin-fixed paraffin-embedded (FFPE) tissues
♦
TFE3 break-apart FISH assay is less prone to fixation problems
t(6;11) renal cell carcinoma
♦
Characterized by a t(6,11)(p21;q12) translocation that results in fusion of the 5′ portion of the alpha gene with the transcription factor gene TFEB at 6p21
Clinical
♦
Less common than Xp11 translocation RCC; fewer than 50 reported to date
♦
Mean and median age, 31 years (range, 9–60 years)
♦
About half of patients present with abdominal pain or hematuria; the rest are asymptomatic
♦
Typically low stage at presentation (T1 or T2)
♦
Majority exhibit indolent biologic behavior
♦
Less than 10% of reported cases have had adverse outcome (metastasis and death from cancer)
Morphology
♦
Typically solid, focal cyst formation, pseudoencapsulated, homogeneous tan–yellow–brown color, without evidence of necrosis
♦
Size ranges from 2 cm to 12 cm in greatest dimension (mean diameter, 7 cm)
♦
Entrapped single native renal tubules characteristically evident at the periphery of the tumor
♦
Solid, nested pattern of growth, biphasic cell population
♦
Predominant cell type is epithelioid, with well-defined borders, abundantly clear, finely granular, or eosinophilic cytoplasm and rounded or vesicular nuclei that have prominent nucleoli evident at low magnification
♦
In all cases, a second cell population comprising 5–30% of the tumor and consisting of smaller cells with denser chromatin, usually clustered around nodules of hyaline basement membrane material (Fig. 35.26)
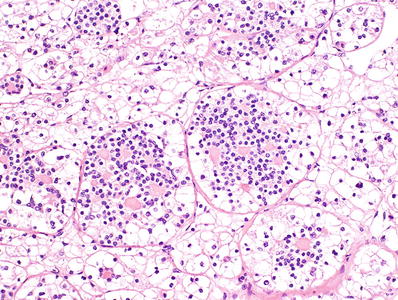
Fig. 35.26.
t(6;11) translocation renal cell carcinoma.
♦
Mitotic figures absent or rare
Immunohistochemistry and FISH
♦
Cytokeratins and EMA are underexpressed
♦
PAX8 consistently positive
♦
Consistently expresses cathepsin K and melanocytic markers Melan A and HMB45
♦
Nuclear immunoreactivity for TFEB is highly specific
♦
TFEB break-apart FISH is the preferred method of confirming the diagnosis in FFPE tissue
Renal Cell Carcinoma Associated with Neuroblastoma
♦
Very rare; fewer than 10 cases
♦
In all instances, renal cell carcinoma arose 3 to 35 years after the child was treated for neuroblastoma at age 2 years or less
♦
Although most were treated with chemotherapy and/or radiation therapy, at least two patients never received either form of treatment, and the etiology of the renal cancer may be on the basis of genetic susceptibility, rather than treatment induced
♦
A few cases of renal cancer after neuroblastoma treatment have been shown to have Xp11 translocation, which has a known association with prior chemotherapy
♦
Tumor size: 1–8 cm; rarely bilateral
♦
Stage at time of diagnosis varies from localized to distantly metastatic
♦
Tumors are heterogeneous, but generally cells are arranged in solid sheets or nests and in papillae, sometimes intermingled with clusters of macrophages and sometimes with psammoma bodies
♦
Tumor cells have sharply defined cell membranes and abundant eosinophilic granular cytoplasm, giving it an “oncocytoid” appearance. In addition, aggregates of cells with abundant reticular or vacuolated cytoplasm are present, with some morphologic resemblance to chromophobe cells (Fig. 35.27)
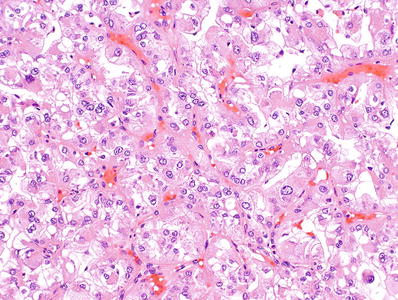
Fig. 35.27.
Renal cell carcinoma associated with neuroblastoma.
♦
Nuclear morphology and grade are variable among individual tumors
♦
Immunoprofile
Positive for EMA, vimentin, and cytokeratins 8 and 18
Negative for cytokeratins 7, 14, 19, and 20
Negative for HMB45, TFE3, TFEB, and S100 protein
♦
Genetics: Clonal abnormalities, none of which are consistent or recurrent
♦
Prognosis: Cases lacking high-grade cytologic atypia have shown indolent behavior. High stage portends a less favorable outcome
Mucinous Tubular and Spindle Cell Carcinoma
Clinical
♦
Rare tumor, less than 1% of renal neoplasms
♦
Originally described as a subset of “low-grade collecting duct carcinoma”
♦
Generally asymptomatic, and most are found incidentally
♦
Female predilection: M:F ratio is about 1:3; mean patient age is 58 years, with a range of 13–82 years
♦
Cytogenetics: Consistent losses involving chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22
♦
Biologic behavior is indolent in most cases
♦
Although rare, recurrence, lymph node metastasis, distant metastasis, and death due to cancer have been reported, in cases with typical bland morphology and in cases with high-grade cytologic features
Macroscopic
♦
Well-circumscribed, gray or light tan, and glistening cut surface (Fig. 35.28)
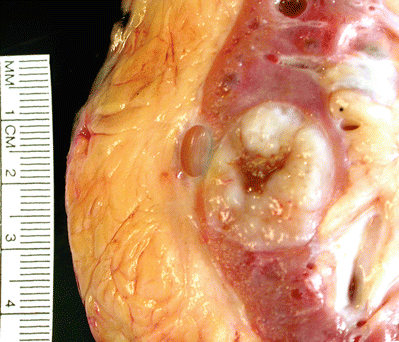
Fig. 35.28.
Mucinous tubular and spindle cell carcinoma. Tumor is a sharply circumscribed gray-white neoplasm with central degeneration and a slightly bulging glistening cut surface.
Microscopic
♦
Tightly packed elongated tubules, arranged in parallel with some branching (Fig. 35.29)
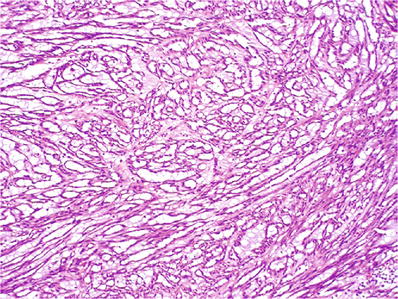
Fig. 35.29.
Mucinous tubular and spindle cell carcinoma. Elongated tubules in a bubbly myxoid stroma consisting of abundant extracellular mucin.
♦
Abundant pale eosinophilic or basophilic extracellular mucin (Fig. 35.30)
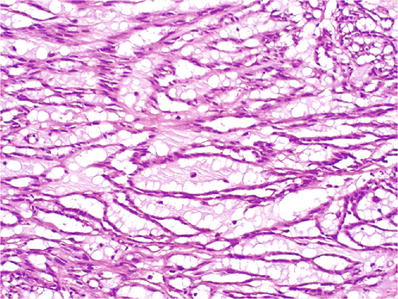
Fig. 35.30.
Mucinous tubular and spindle cell carcinoma. Higher power view of the tumor shown in Fig. 35.42, illustrating the extremely bland nuclear characteristics of the tumor, possibly accounting for its very indolent behavior.
♦
Collapsed tubules may impart a spindled appearance
♦
Tumor cells are usually cytologically low grade, round or oval, although rare cases with high-grade/sarcomatoid features have been reported
Immunohistochemistry
♦
Most express CK7 and AMACR
Tubulocystic Renal Cell Carcinoma
Clinical
♦
A rare tumor, less than 1% of renal carcinomas
♦
Originally described as a subset of “low-grade collecting duct carcinoma”; assessment of cell of origin presently under investigation
♦
Two-thirds of patients are asymptomatic, and the tumor is found incidentally
♦
Male to female ratio is about 6:1; mean patient age is 58 years with a range of 30–94 years
♦
Cytogenetics: Gains of 7 and 17 and loss of Y
♦
Tumor is relatively indolent; survival rate exceeds 90%; one recurrence and four instances of metastasis in 70 reported cases
♦
Tumors are generally low stage when initially detected
Macroscopic
♦
Well circumscribed, with a mean size of 4.2 cm (range, 0.7–17 cm)
♦
Spongy “bubble wrap-like” cut surface; grossly composed of innumerable cysts of variable size
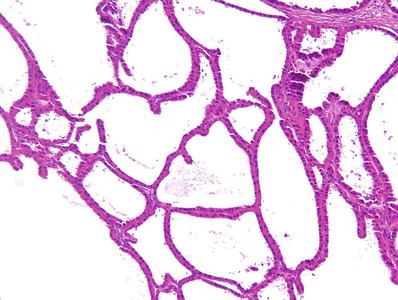
Microscopic
♦
Cystically dilated tubules of variable size, separated by thin “spidery” septa (Fig. 35.31) and lined by a single layer of flat, cuboidal, or hobnail cells (Fig. 35.32)