CHAPTER 36 Natural anticoagulants and thrombophilia
Coagulation
Classic cascade mechanism of coagulation vs. cell-based models of thrombin generation
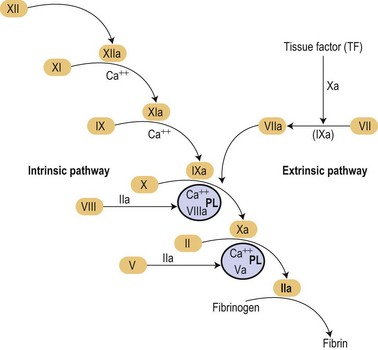
In the mid 1960s two different investigators conceived an original concept of the coagulation mechanisms triggered as a ‘cascade’ of proteolytic reactions acting as biological amplifiers.1,2 According to those classic concepts, coagulation mechanisms were activated through intrinsic and extrinsic pathways converging on a common pathway that will finally activate prothrombin into thrombin that would cause soluble fibrinogen to become polymerized into a solid fibrin clot (Fig. 36.1).
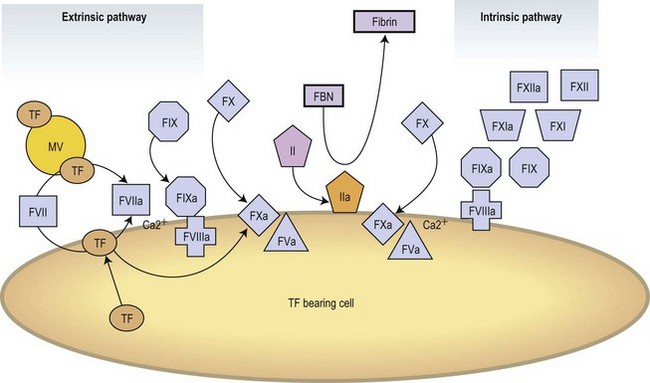
Modern concepts have integrated the classic stepwise cascade into a more comprehensive scheme in which blood coagulation is initiated by cellular components in vivo.3 Actual models contemplate the implication of cellular and enzymatic mechanisms in three differentiated steps: Initial activation, propagation and thrombin generation (Fig. 36.2). Fibroblasts, smooth muscle cells of the vascular wall, activated monocytes or endothelial cells, or even platelets are potential cellular sources of TF.4,5 According to current knowledge, the activation of the coagulation is initiated at the very moment in which TF is exposed on cellular surfaces. TF is a transmembrane protein that binds plasma factor VII/VIIa. In a first step, the TF exposed on damaged vascular areas forms a complex with FVII/FVIIa. FVIIa is present in reduced amounts in circulating blood. The TF : FVIIa complexes will activate FIX and FX. FXa generated will activate small quantities of prothrombin (FII) into thrombin (FIIa). Small amounts of thrombin generated in this short loop will be able to promote further activation of the coagulation by further activating FV and FVIII in the presence of platelet phospholipids and FIX.3 During the propagation step FIXa bound to FVIIa on the surface of activated platelets form the tenase complex that will further activate FX into FXa. FXa binds to FVa on the phospholipid surface provided by activated platelets and form the prothrombinase complex. During the final step of the cell-based model, the prothrombinase complex will further magnify the generation of thrombin facilitating the conversion of prothrombin into thrombin. Large amounts of thrombin generated during the final step will convert fibrinogen into fibrin. Thrombin itself activates FXIII thus stabilizing the fibrin formed.
Molecular mechanism
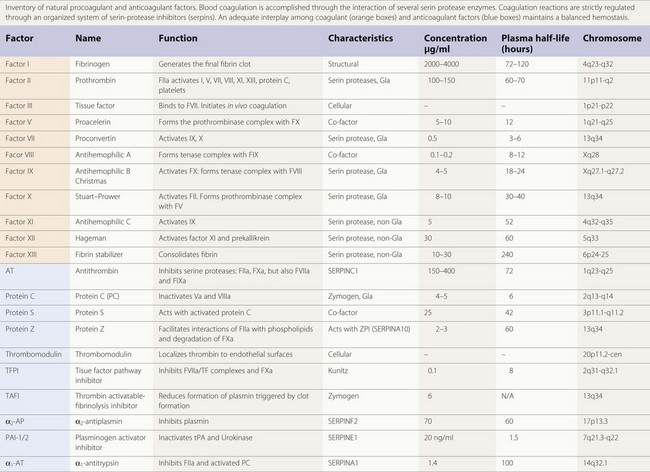
The majority of enzymes in the coagulation cascade are members of the same group of proteins called serine proteases (Table 36.1). They exhibit their function by limited proteolysis of peptide bounds of their substrates. The cascade design of the coagulation system allows a great amplification of signal in each step, since the enzymes activate more than one molecule of substrate. However, the enzymes by themselves do not have high catalytic efficiency and therefore the assembly of enzyme complexes is of crucial importance. This is for instance demonstrated by the bleeding diathesis experienced by hemophilia A patients, where the co-factor FVIII is deficient. In the enzyme complexes the activated membrane, exposing negatively charged phosphatidyl-serine plays a key role. Both the substrate and enzyme bind to the membrane concentrating the proteins at the surface. The correct orientation of the proteins on the cell surface facilitates the interaction. The importance of the membrane is shown in studies of the prothrombinase complex where the addition of phospholipid membrane gives an about 1000-fold increase in catalytic efficiency. Addition of co-factor FVa gives an additional increase and thus the prothrombinase complex is 106 times more efficient than the enzyme alone.6
Table 36.1 Procoagulant and anticoagulant factors
 |
Vitamin K-dependent proteins and their binding to the membrane
The vitamin K dependent plasma proteins are a group of Gla domain containing proteins that require vitamin K for their synthesis7 (Table 36.1). The Gla domains contain 9–12 Glu residues that are γ-carboxylated in a post-translational process that require vitamin K. The γ-carboxylated Glu residues (Gla residues) mediate the binding of Ca2+ ions and are crucial for the membrane interaction. Thus, in patients on treatment with vitamin K antagonist (coumarin) the γ-carboxylation is impaired leading to loss of calcium and membrane binding. Due to high sequence similarity, the fold and phospholipid binding mechanism are thought to be similar for all the Gla domains. Although the sequence of the Gla domains are very similar for all the vitamin K zymogens, their affinities for the membrane surface vary 100 to 1000-fold. It has been suggested that this is caused by amino acid sequence differences in just a few positions.8
Regulatory mechanisms
Activation of coagulation switches on a series of mechanisms aimed at controlling and correcting fibrin formation. Plasminogen is converted into plasmin to initiate fibrinolysis. Mechanisms of coagulation and fibrinolysis are regulated at different steps by inhibitory mechanisms that maintain an adequate balance. This chapter is mainly focused on the implication of natural anticoagulants in thrombophilia. Certainly genetic or acquired alterations of fibrinolysis may result in delayed cleavage of the fibrin clot formed resulting in an indirect hypercoagulable state or in persistency of the associated symptoms.9,10 Despite the critical role of the fibrinolytic system in the resolution of thrombotic events, the overall impression is that its contribution to thrombophilia is relatively low when compared with deficiencies of natural anticoagulants.
SERPINs: a family of coagulation inhibitors
An important proportion of the natural anticoagulants exert their inhibitory action through inhibition of the enzymatic action of procoagulant serine proteases. This family of inhibitory proteins is called SERPINs (SERine Protease INhibitors). Antithrombin (SERPINC1), heparin co-factor II (SERPIND1), protein C inhibitor (SERPINA5) and PAI-1 (SERPINE1), are representative members of this family of inhibitors (Table 36.1). Although their primary sequence similarity is low, these proteins have a conserved three-dimensional structure that is crucial for their function in the regulation of hemostasis, thrombosis and fibrinolysis.11 Their mechanism of action can be compared to a mousetrap, where the serine protease to be inhibited is lured into an irreversible binding leading to a distortion of the enzyme and loss of activity.12 The inhibition is irreversible and one molecule of serpin can only inhibit one molecule of serine protease.
Antithrombin
Antithrombin (AT) is a α2-globulin, member of the serpin family of protease inhibitors (SERPINC1), synthesized in the liver and is the major inhibitor of blood coagulation. Human AT is a single chain glycoprotein of 432 aminoacids with a reactive site cleaved by target enzymes located at Arg393-Ser 394. Antithrombin is a very powerful inactivator of thrombin (FIIa) and FXa, but does also inhibit FVIIa, FIXa, FXIa and FXIIa to a lesser extent.13 Antithrombin has a molecular weight of 58 200 kDa. There are two isoforms, α and β, of circulating AT. The α represents 90–95% and the β, 5–10% of the total. Under physiological conditions AT circulates in a form that expresses low inhibitory affinity. AT possess a binding site for heparin, being the β isoform the fraction with a higher affinity for heparin.14 Under physiological conditions heparin sulfates present on the surface of endothelium will provide a background level of activation on AT. Interaction of specific glycosaminoglycan sequences with the specific heparin-binding domain induces a conformational change in AT and accelerates by an order of a thousand times its inhibitory action on activated serin proteases.15 Inactivation of thrombin (FIIa) by AT produces thrombin–antithrombin complexes (TAT) that can be determined and used as a surrogate marker of the activation of the coagulation. The anticoagulant action of conventional heparins, low molecular weight heparins, and pentasaccharide is mediated by AT. Congenital or acquired deficiencies of AT, may reduce the therapeutic action of the previous anticoagulants. In addition to its anticoagulant action AT has an important anti-inflammatory effect that seems closely dependent on its ability to bind to endothelial glycosaminoglycans.
Protein C system
Protein C structure
Protein C is a vitamin K-dependent protein of 62 kDa that circulates in blood as a zymogen in a concentration of about 4 µg/ml. The protein is synthesized in the liver. The mature protein C molecule is composed of a light and a heavy chain, the two chains being disulfide-linked. The light chain consists of a Gla domain and two epidermal growth factor (EGF)-like domains. The heavy chain contains a short activation peptide and a serine protease domain. Similar to the procoagulant co-factors, protein C circulates as an inactive proenzyme and needs to be activated to gain anticoagulant activity. Like for the other vitamin K-dependent proteins, the Gla domain of protein C mediates the membrane binding. Compared to the other vitamin K-dependent coagulation proteins, protein C has a quite low affinity for the membrane.8
Protein C activation complex
Protein C is activated by thrombin in complex with thrombomodulin.16 Thrombomodulin is a transmembrane glycoprotein consisting of a short cytoplasmic tail, a well-conserved membrane spanning region and a serine-threonine rich domain followed by 6 EGF-like repeats.17 Upon binding to thrombomodulin, thrombin changes its substrate specificity and is able to activate protein C. The efficiency of the thrombomodulin/thrombin complex is low and an additional co-factor, the endothelial receptor EPCR is needed for fully efficient protein C activation.
Protein C function
Activated protein C (APC) down-regulates the blood coagulation by limited proteolytic cleavage of co-factors FVIIIa and FVa. FVa and FVIIIa are homologous co-factors with the same domain structure: A1-A2 (the heavy chain), the B domain (only present in the non-activated FV and FVIII) and A3-C1-C2 (the light chain). APC cleaves at sites in the heavy chains of the proteins, at Arg306, Arg506 and Arg679 in FVa and Arg336 and Arg562 in FVIIIa.18 For the inactivation of FVa, the Arg506 cleavage is kinetically favored, but leads only to a partial loss of pro-coagulant function, due to decreased affinity for FXa. For complete loss of activity the cleavage at Arg306 is also needed, which leads to dissociation of the fragments. The FVIIIa is by itself an unstable molecule and is spontaneously degraded by dissociation of the A2 domain. Cleavage of FVIIIa at Arg336 is kinetically favored over cleavage at Arg562 in APC-mediated inactivation of FVIIIa. The cleavage at Arg336 does however not lead to complete loss of activity and the additional cleavage at Arg562 is needed.19
Protein S
Protein S is a 70 kDa large vitamin K-dependent protein that circulates in the blood in a concentration of 20–25 mg/ml. More than 60% of the protein S circulates in blood bound to a complement regulator protein called C4b-binding protein (C4BP).20 Protein S is mainly synthesized in the liver but synthesis has also been observed in endothelial cells, by testicular Leydig cells and by osteoclasts.21 Protein S has the following domain structure: an N-terminal vitamin K-dependent Gla domain, a thrombin-sensitive region (TSR), four EGF-like domains and two laminin G-type (LamG) domains (also called the SHBG-like domain). Like the other vitamin K-dependent blood proteins, the Gla domain of protein S mediates the membrane binding. Of the vitamin K-dependent proteins, protein S has the highest affinity for the membrane surface.8 The TSR is not found in the other vitamin K-dependent proteins. The region is sensitive to thrombin cleavage and cleavage leads to loss of APC co-factor function.22 Factor Xa has also been reported to cleave in the TSR, with the same result.
Protein S function
Protein S is an important co-factor for APC both in the inactivation of FVa and FVIIIa. In the FVa-inactivation protein S stimulates the proteolytic cleavage at Arg306 about 20-fold and only minor stimulation of the Arg506-cleavage is observed.18 However, when FVa participates in the prothrombinase complex protein S also influences the cleavage at Arg506 by counteracting the FXa-mediated inhibition of this cleavage site. It was originally thought that only the free form of protein S stimulates the APC-mediated cleavage of FVa; however, a recent report indicates that protein S in complex with C4BP can also stimulate the cleavage at Arg306.23 In the inactivation of FVIIIa protein S functions as a co-factor synergistically with the intact form of factor V.18 Protein S stimulates both the cleavage sites in FVIIIa.
In addition to being a co-factor for APC, protein S has been shown to exhibit an APC-independent anticoagulant activity.24 It has been demonstrated that the APC-independent anticoagulant function of protein S is an effect on the TFPI inhibition of the extrinsic pathway.25 As there is covariation of plasma TFPI and protein S levels both in health and in disease, these findings suggest that the risk of DVT associated with protein S deficiency states might be in part explained by the accompanying low plasma TFPI levels.24
Tissue factor pathway inhibitor TFPI
TFPI, also known as lipoprotein-associated coagulation inhibitor or extrinsic pathway inhibitor, is a Kunitz-type inhibitor with a molecular weight of 34 kD.26 The molecule possesses an acidic amino terminus, three Kunitz-type domains and a basic carboxy terminus. Circulating TFPI is synthesized by endothelial cells and smooth muscle cells.27 Approximately 10% of the blood TFPI is present in platelets. TFPI neutralizes FVIIa-TF complexes, but is also capable of neutralizing FVIIa and FXa, though at a much slower rate. In fact, the inhibitory action of TFPI on FVIIa-TF complexes requires certain levels of FXa. In experimental animals, administration of TFPI reduces mortality in a model of septic shock induced by Escherichia coli.28
Abnormal levels of TFPI or mutation in the respective gene are not strongly associated with thrombophilia. Treatment with exogenous hormones has a profound lowering effect on TFPI levels in women, with lower levels in oral contraceptive users than in premenopausal non-users. Levels of TFPI are strongly reduced levels in patients with abetalipoproteinemia, though they do not seem to be exposed to an enhanced risk of thrombosis. Recent studies indicate that low levels of TFPI, especially low TFPI-free and total antigen in plasma, may constitute a risk factor for DVT.29
Protein Z-dependent protease inhibitor
Protein Z is a vitamin K-dependent protein with a molecular weight of 62 kD, plasma concentration ranging from 2 to 3 µg/ml and with an approximate half-life of 2.5 days. Although the structure of protein Z contains a Gla domain it does not possess protease activating action on other coagulation factors. Protein Z promotes FXa inhibition induced by ZPI.30 Protein Z-dependent protease inhibitor (ZPI) is another member of the serpin family (SERPINA10) with known inhibitory action on FXa and FXIa.31 Heparin enhances the interaction of ZPI with FXIa.
The prothrombotic phenotype associated to protein Z-ZPI deficiency is controversial. Knock out mice for the protein Z gene do not seem to develop a thrombotic tendency unless there is another associated condition such as factor V Leiden.32 It has been postulated that thrombotic potential of the protein Z and ZPI deficiencies studies in humans question the thrombotic relevance of both ZPI and PZ deficiencies except when they act in combination with other prothrombotic factors.30
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


