CHAPTER 25 Myeloproliferative neoplasms with eosinophilia
In the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues1 the term ‘myeloproliferative neoplasm’ (MPN) is preferrred to myeloproliferative disorder. Within the group of MPN is chronic eosinophilic leukemia, not otherwise specified.2 Other neoplasms in which eosinophils constitute a dominant or major component are categorized according to the underlying molecular defect, a fusion gene incorporating part of PDGFRA, PDGFRB or FGFR1.3 In the first and last of these groups of disorders there can also be a lymphoid component to the neoplasm, indicating that the responsible mutation occurred in a pluripotent lymphoid-myeloid stem cell. There are other MPN in which eosinophils are part of the neoplastic population but they are not usually a dominant feature. These include chronic myelogenous leukemia (BCR-ABL1 positive) and systemic mastocytosis (SM). In addition, other well-defined MPN can undergo an eosinophilic transformation. The WHO categories of MPN with eosinophilia and a well defined genetic abnormality will be discussed first since a diagnosis of eosinophilic leukemia, not otherwise specified, can be made only when they have been excluded. Non-neoplastic disorders with eosinophilia are discussed in Chapter 16.
Myeloid and lymphoid neoplasms with PDGFRA rearrangement
Most neoplasms resulting from rearrangement of PDGFRA present as chronic eosophilic leukemia (CEL).3,4 The natural history of this condition includes blastic transformation including myeloid sarcoma. A minority of patients present with acute leukemia, either myeloid or T-lymphoblastic with preceding eosinophilia being documented or being present at the time of diagnosis.5
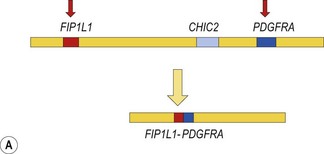
The prototype of this group of neoplasms is CEL associated with a FIP1L1-PDGFRA fusion gene that has resulted from a cryptic interstitial deletion at 4q12. Until the discovery of this recurrent molecular abnormality6 these patients were regarded as having the idiopathic hypereosinophilic syndrome (HES). The diagnosis of idiopathic HES can now not be made without exclusion of FIP1L1-PDGFRA.
Clinicopathological features result from tissue infiltration by eosinophils and organ damage by release of eosinophil granule contents (Table 25.1). Endocardial and myocardial damage is prominent, as is thrombosis.
Table 25.1 Possible clinicopathological features of chronic eosinophilic leukemias
| Organ or system | Features |
|---|---|
| General | Fatigue, fever, weight loss, night sweats, headache |
| Hematological | Eosinophilia (by definition), variable increase in other leukocytes and their precursors, anemia, thrombocytopenia, increased bone marrow mast cells (in some subtypes), increased bone marrow reticulin, clonal cytogenetic or molecular abnormalities, increased serum vitamin B12, increased serum tryptase (in some subtypes) |
| Spleen | Splenomegaly, splenic infarction |
| Liver | Hepatomegaly |
| Lymph nodes | Lymphadenopathy (some subtypes) |
| Cardiovascular | Endocardial, myocardial and pericardial infiltration and damage, increased serum troponin, restrictive cardiomegaly, dilated cardiomyopathy, valvular incompetence, intracardiac thrombi and systemic embolization, cardiac failure, vasculitis, arterial thrombosis, Raynaud’s phenomenon, digital necrosis, venous thromboembolism and pulmonary embolism |
| Respiratory | Cough, dyspnea as a result of infiltration, restrictive or obstructive pulmonary disease, pleural effusion |
| Skin | Pruritis, urticaria, angioedema, papules or nodules due to infiltration, vasculitis |
| Central and peripheral nervous system | Cranial nerve palsies, paraspinal masses, cerebral embolism, peripheral neuropathy |
| Gastrointestinal | Diarrhea as a result of infiltration, malabsorption, mucosal ulcers |
| Musculoskeletal | Myalgia, bone pain |
Precise diagnosis of this condition is very important because of its sensitivity to treatment with imatinib and other tyrosine kinase inhibitors (TKI). Even patients who present in acute transformation can respond to imatinib. Because of the possibility of cardiac damage, rapid diagnosis can also be important. An algorithm showing a diagnostic approach to suspected eosinophilic leukemia is shown in Fig. 25.1.
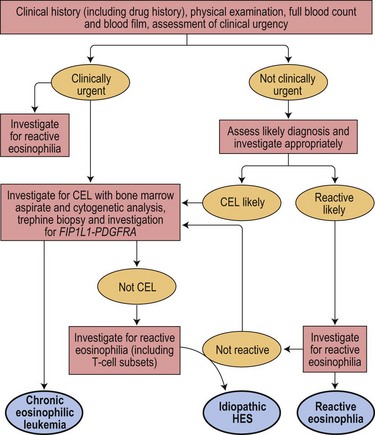
Fig. 25.1 An algorithm showing a diagnostic approach to eosinophilia and suspected eosinophilic leukemia
Pathologic features
Peripheral blood
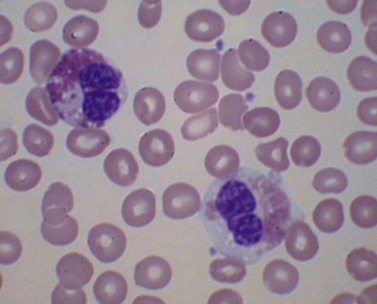
There is eosinophilia and often neutrophilia. Some patients are anemic or thrombocytopenic. The eosinophils may be cytologically normal but often there is degranulation, cytoplasmic vacuolation or nuclear abnormalities – either lack of segmentation or hypersegmentation (Fig. 25.2). Eosinophils may be sufficiently abnormal that they are not recognized by automated blood analyzers.
Bone marrow
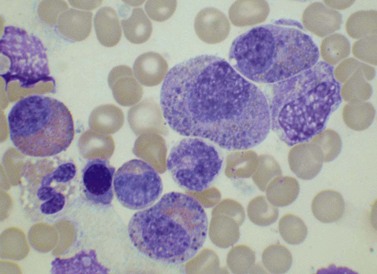
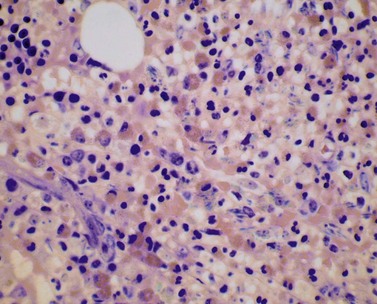
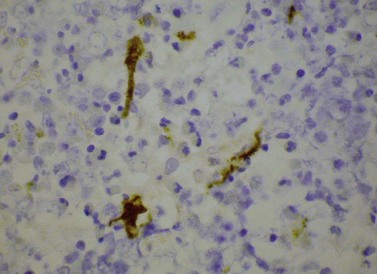
The bone marrow (BM) aspirate is hypercellular as the result of an increase in eosinophils and their precursors. Eosinophil precursors may have purple-staining proeosinophilic granules (Fig. 25.3). There may also be an increase in neutrophils and precursors. Bone marrow trephine biopsy (BMTB) sections show dominance of granulopoiesis with prominent eosinophilia (Fig. 25.4). Reticulin is increased. The majority of patients also have increased mast cells (Fig. 25.5). These are often spindle-shaped and usually dispersed or in loose clusters but in occasional patients they form cohesive infiltrates similar to those of SM. The mast cells can express CD25, an abnormality that is also a feature of SM; they may or may not express CD2, which is usually expressed in SM.3,7 CEL with increased BM mast cells has sometimes been misinterpreted as SM but it must be appreciated that SM with a KIT mutation and CEL with rearrangement of PDGFRA are distinct and different diseases.
Supplementary investigations
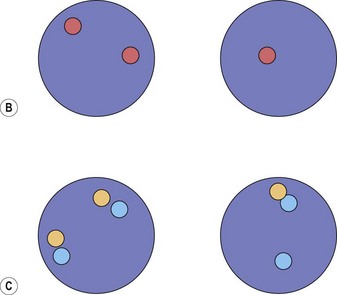
Cytogenetic analysis is usually normal but a minority of patients have a translocation with a 4q12 breakpoint or an unrelated abnormality such as trisomy 8. A secondary cytogenetic abnormality such as trisomy 8 may appear for the first time at evolution to AML.8 Diagnosis is dependent on molecular analysis or fluorescence in situ hydridization (FISH) analysis. The reverse transcriptase polymerase chain reaction (RT-PCR) may demonstrate the FIP1L1-PDGFRA fusion gene but sometimes RT-PCR is negative and nested RT-PCR is required for demonstration of the fusion gene. Various FISH techniques are used. Mainly they rely on demonstration of deletion of the CHIC2 gene, which is located between FIP1L1 and PDGFRA and is thus included in the cryptic deletion (Fig. 25.6).9 Probes flanking FIP1L1 and PDGFRA can also be used with a dual-color break-apart technique.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


