CHAPTER 20 Myelodysplastic syndromes
The first report of a patient with myelodysplastic syndrome (MDS) dates from 1900, when Leube described a patient with severe megaloblastic anemia preceding development of overt leukemia.1 This description was followed by several reports of patients characterized by cytopenia, disturbed maturation of bone marrow (BM) precursors, an increase of blasts and a significant risk of evolving to acute myeloid leukemia (AML).1 The first international classification of MDS was developed by the French-American-British (FAB) Group2 (Table 20.1) and extended by the World Health Organization (WHO) 2001 and WHO 2008 (Table 20.2) classifications.3–5 The diagnosis of MDS is an effort that requires clinical, cytogenetic and pathological expertise, and often also repeated BM studies.
Table 20.1 French-American-British (FAB) group classification of myelodysplastic syndromes2
| Type | Peripheral blood | Bone marrow |
|---|---|---|
| RA | <1% blasts | Dyshemopoiesis in one, two or all three lineages; <5% blasts |
| RARS | <1% blasts | As RA with ring sideroblasts (RS) comprising >15% erythroblasts |
| RAEB | <20% blasts | As RA with 5–20% blasts in BM, +/− RS |
| RAEBt | >30% blasts | As RA with 20–30%* blasts in BM or as RAEB with Auer rods |
| CMML | As any of the above plus >1 × 109/l monocytes/promonocytes in blood | |
RA, refractory anemia; RARS, RA with ring sideroblasts; RAEB, RA with excess of blasts; RAEBt, RAEB in transformation; CMML, chronic myelomonocytic leukemia.
Variants that do not fit well into this classification include hypoplastic MDS, fibrotic MDS and juvenile myelomonocytic leukemia.
* Cases with >30% blasts were classified as acute leukemia.
Table 20.2 WHO 2008 classification of myelodysplastic syndromes4
| Disease | Blood findings | Bone marrow findings |
|---|---|---|
| Refractory cytopenia with unilineage dysplasia (RCUD): | Unicytopenia or bicytopenia1 | Unilineage dysplasia: ≥10% of the cells in one myeloid lineage |
| [Refractory anemia (RA); refractory neutropenia (RN); | No or rare blasts (<1%)2 | <5% blasts |
| Refractory thrombocytopenia (RT)] | <15% of erythroid precursors are ring sideroblasts | |
| Refractory anemia with ring sideroblasts (RARS) | ||
| Refractory cytopenia with multilineage dysplasia (RCMD) | ||
| Refractory anemia with excess blasts-1 (RAEB-1) | ||
| Refractory anemia with excess blasts-2 (RAEB-2) | ||
| Myelodysplastic syndrome – unclassified (MDS-U) | ||
| MDS associated with isolated del(5q) |
Although the finding of 5–19% blasts in the blood is, in itself, diagnostic of RAEB-2, cases of RAEB-2 may have less than 5% blasts in the blood if they have Auer rods and/or 10–19% blasts in the marrow. Similarly, cases of RAEB-2 may have less than 10% blasts in the marrow but may be diagnosed by the other two findings, Auer rod+ and/or 5–19% blasts in the blood.
1 Bicytopenia may occasionally be observed. Cases with pancytopenia should be classified as MDS-U.
2 If the marrow myeloblast percentage is less than 5% but there are 2–4% myeloblasts in the blood, the diagnostic classification is RAEB 1. Cases of RCUD and RCMD with 1% myeloblasts in the blood should be classified as MDS-U.
3 Cases with Auer rods and <5% myeloblasts in the blood and <10% in the marrow should be classified as RAEB-2.
Definition
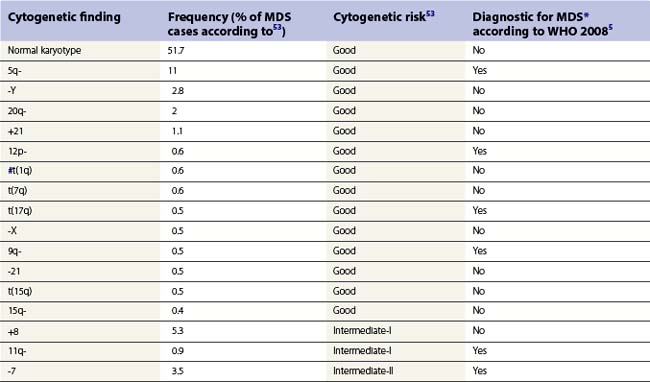
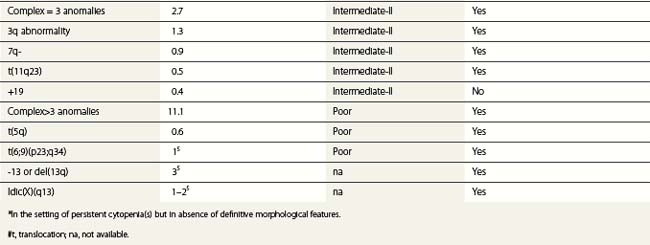
According to the WHO 2008 classification, MDS may be defined as a group of clonal hematopoeitic stem cell disorders characterized by cytopenia(s), dysplasia in one or more of the major myeloid cell lines, ineffective hematopoiesis and increased risk of AML development.5 The BM is usually hypercellular. Myelodysplasia, i.e. abnormal maturation in hematopoiesis, is a hallmark of MDS (Table 20.3). However, myelodysplasia is not definitive evidence of MDS and may be present as a result of various nutritional, toxic, infectious and other factors (Table 20.4). Moreover, a presumptive diagnosis of MDS may be made in a patient with unexplained cytopenia(s) in the absence of overt myelodysplasia if certain defining cytogenetic abnormalities are present (Table 20.5).
Table 20.3 Morphologic signs of dysplasia in blood and bone marrow cells in MDS
| Lineage | Blood | Bone marrow |
|---|---|---|
| Erythropoiesis | Macrocytes Aniso-poikilocytosis Dimorphic picture Polychromasia Punctate basophilia Normoblasts Reticulocytopenia | Erythroid hypercellularity Multinuclearity Dyskaryorrhexis Megaloblasts Cytoplasmic vacuoles Howell–Jolly bodies Ring sideroblasts |
| Granulopoiesis | Hypogranular neutrophils Unilobed or bilobed neutrophils (Pelger cells) Hypersegmented neutrophils Monocytosis (often with multiple, elongated nuclear lobes) Promonocytes (with fine azurophil granules) Degranulated eosinophils | Hypogranularity of myeloid precursors Increased promonocytes Increased blast cells (type I with scanty agranular cytoplasm and type II with sparse granules) |
| Thrombopoiesis | Agranular platelets | Micromegakaryocytes |
| Giant platelets | Large megakaryocytes with single round or oval nucleus | |
| Megakaryocyte fragments | Large megakaryocytes with multiple small round nuclei Megakaryoblasts |
Table 20.4 Non-clonal causes of dysplasia
| Dyserythropoiesis |
| Dysgranulopoiesis |
| Dysmegakaryocytopoiesis |
Epidemiology
American studies report the median age of MDS patients at diagnosis as 76 years.6 In Asian countries the median age at diagnosis is 10 years lower.7 The incidence is higher in men than in women (4.5 vs 2.7 cases/100 000 population/year, respectively). Incidence increases with age with 86% of MDS patients being older than 60 years of age. A prevalence of 1 in 500 in the population over 60 years of age has been suggested.8 In patients over 85 years, MDS represents about a quarter of all hematologic malignancies. Although studies point towards an increasing incidence, this may at least in part reflect a greater willingness to perform BM investigations in the elderly and increased reporting.9
Etiology of primary MDS is still poorly understood. Certain genetic disorders and exposure to pesticides, organic solvents or ionizing radiation have been implicated as risk factors.10,11 A large prospective study indicated obesity and smoking as modifiable, although weak, risk factors, while intake of alcohol, meat, fruit and vegetables, and physical activity did not influence the risk of MDS.12 Secondary MDS, which tends to occur in younger people, develops after exposure to certain chemicals and cytotoxic drugs, especially alkylating agents.13,14 The younger age at presentation of idiopathic MDS in developing countries suggests a less rigorous control of noxious chemicals in these communities.15 Childhood MDS is very rare (1.8 cases/1 000 000 population/year).16 Familial MDS is also very rare and occurs at a younger age.17
Classification
The ‘minimal’ morphologic criteria for MDS diagnosis according to WHO classification is the presence of at least 10% dysplastic forms in at least one hematopoietic lineage (erythroid, granulocytic or megakaryocytic), in an appropriate clinical setting.4 The FAB classification (Table 20.1) applied the arbitrary limit of 30% blasts for diagnosis of AML, while this limit has been changed to 20% in the WHO classification abolishing the former category of RAEBt. There is still ongoing debate as to how patients with a blast count between 20% and 30% should be classified.4 Of note, patients with specific translocations such as t(8;21), inv.16 and t(15;17) are classified as AML even if numbers of blasts are below 20%. This rule does not apply to cases of AML with t(9;11)(p22;q23), t(6;9)(p23;q34), inv.3(3)(q21q26.2), t(3;3)(q21;q26.2) and t(1;22)p(13;q13) that are considered as MDS if the number of blasts does not reach 20% within 2 months’ follow-up time. Some AML cases may be recognized as myelodysplasia-related, which should be stated in the report (see Chapter 18). Some of these cases would have been previously diagnosed as RAEB in transformation (RAEBt) and some would have been called AML. It is probable that a large proportion of AML cases in the elderly will fall into AML myelodysplasia-related category.
The 2001 WHO classification took into consideration the extent of dysplasia (unilineage vs multilineage), which has been proven to be of prognostic significance.18,19 WHO 2008 (Table 20.2) extended the definition of cases with unilineage dysplasia, adding two other categories to refractory anemia (RA): refractory neutropenia (RN) and refractory thrombocytopenia (RT). In WHO 2008, the presence of ring sideroblasts is only of significance in the context of unilineage erythroid dysplasia (RA vs RA with ring sideroblasts, RA-RS). Patients with multilineage dysplasia and less than 5% blasts are diagnosed as refractory cytopenia with multilineage dysplasia (RCMD) independent of the presence of sideroblasts. Refractory anemia with excess of blasts (RAEB) is separated into RAEB I with 5–9% blast cells and RAEB II with 10–19% blast cells in BM smears. WHO 2008 has also stressed the significance of the presence of blasts in peripheral blood (PB), since patients with 2–4% PB blasts and <5% BM blasts are now categorized as RAEB-1.4 It is important to note that most of these qualitative and quantitative parameters have been derived from blood and aspirated BM appearances. Correlation with their assessment by BM trephine biopsy (BMTB) histology has only a limited evidence base.
The revised WHO classification of 2008 has also recognized differences between MDS in adults and in children. The latter usually present with leukopenia and/or thrombocytopenia and less often with anemia. A provisional entity ‘refractory cytopenia of childhood’ (RCC) has been introduced, which includes childhood presentations with multilineage dysplasia, <2% blasts in PB, <5% blasts in BM and persistent cytopenia. Childhood MDS patients who have increased numbers of blasts should be classified in the same way as adults.4,5
Diagnosis
Symptoms of anemia (tiredness, breathlessness and lassitude), thrombocytopenia (bruising or bleeding), or neutropenia (recurrent infections, mouth ulcers) are most common. A large fraction of patients with MDS are asymptomatic and may be diagnosed incidentally when a blood test, performed for an unrelated reason, shows macrocytic anemia. Extramedullary involvement (such as lymphadenopathy and/or splenomegaly) is relatively rare and possibility of MDS/MPN should be first excluded in these patients. Approximately 10% of patients display autoimmune symptoms such as vasculitis, arthritis, polyneuropathy and connective tissue disease-like presentations. The tumor suppressor gene interferon regulatory factor-1 (IRF-1) has been implicated in the pathophysiology of MDS-associated autoimmune deregulation.20
A full blood count may reveal anemia, neutropenia, thrombocytopenia or multiple cytopenias. The recommended level for defining anemia is hemoglobin <10 g/l. Neutropenia is defined by absolute neutrophil count <1.8 × 109/l and thrombocytopenia by platelet count <100 × 109/l. A raised mean cell volume (MCV) is frequently found and may be the only abnormality in the blood count. However, anemia may also be normocytic or microcytic. There is almost always reticulocytopenia. In patients with bone marrow fibrosis, a leukoerythroblastic picture may be found. In cases with del(5q), there may be a slightly elevated platelet count. Other tests are usually normal, but in some patients increased levels of lactate dehydrogenase (LDH) may be present, as a result of increased cell death in BM. High LDH levels carry a poor prognosis. Serum B12 levels are usually high, but coincident pernicious anemia has also been reported.21 A positive direct antiglobulin test, with signs of hemolysis, is found in 8% of patients.22 Both leukocyte and platelet function tests may be impaired,23,24 as well as the growth of BM progenitor cells in short-term and long-term cultures.25 Recent studies stress decreased numbers of early multilineage precursors and erythroid progenitors as characteristics of MDS.26
Diagnosis of MDS depends on BM examination and cytogenetics. However, some patients with persistent mild cytopenia and/or mild dysplasia do not fulfill minimal criteria for MDS and should be followed. For these patients, the terms idiopathic cytopenia of undetermined significance (ICUS) or idiopathic dysplasia of undetermined significance (IDUS) may be applied;27 some of these patients will, with time, develop MDS.
Blood film examination
Blood films should be prepared fresh since blood samples exposed to anticoagulants are unsatisfactory for accurate morphology. The ability to diagnose MDS depends crucially on optimal staining of PB and BM films. Staining varies enormously between laboratories, so observers should become familiar with their own laboratory’s stains. Films sent for a second opinion should be sent unstained. Blood films are most commonly stained using the May–Grünewald–Giemsa or Wright method.28 It is important that the stain used picks up granularity in neutrophils well and also reveals basophilic stippling in red cells.
Erythrocytes
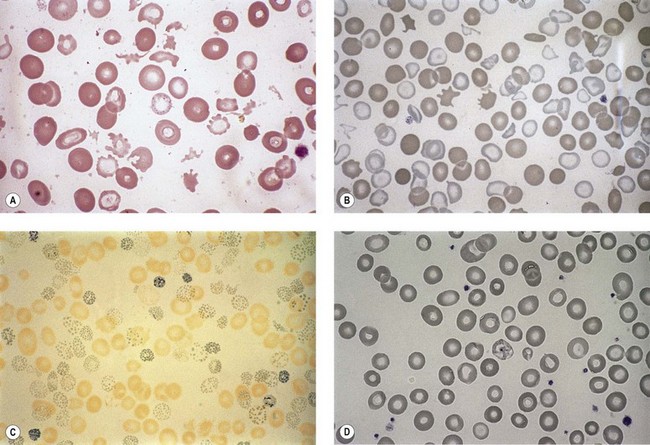
Anemia is the most common feature of MDS but it is not an obligatory finding. Usually the red cells are large, but they may be of normal size and occasionally are small. Many textbooks list sideroblastic anemia among the causes of a low MCV but this may be misleading, since it only applies to the rare hereditary form. The MDS category of refractory anemia with ring sideroblasts (RA-RS) is associated with a raised or normal MCV. Frequently, red cells in MDS vary in size and shape (aniso-/poikilocytosis). Oval macrocytes are common. When these are accompanied by small hypochromic cells, giving a dimorphic picture, RA-RS should be suspected (Fig. 20.1A). However, small hypochromic red cell fragments may be seen in all forms of MDS. A preponderance of small hypochromic red cells is rarely seen and should suggest acquired hemoglobin H (HbH) disease (Fig. 20.1B). This is a very rare finding in MDS and may be revealed with supravital staining (Fig. 20.1C).29 Poikilocytosis refers to several types of shape changes: dacrocytes (tear-drop shaped cells), acanthocytes (spur-spike cells), and cell fragments are all frequently seen. MDS is a rare cause of elliptocytosis (small, oval erythrocytes). However, red cell size and shape may be normal. Howell–Jolly bodies are occasionally seen but rather more common is basophilic stippling, which may be fine or coarse (Fig. 20.1D). Fine basophilic stippling confined to large, misshapen, hypochromic cells usually indicates RA-RS.
Leukocytes
Neutropenia is very common in MDS. Although it may already be severe at diagnosis, it is frequently mild or may develop later in the course of the disease. Abnormalities in granularity of neutrophils are important features. Classically, there are few if any neutrophil granules (Fig. 20.2A) but sometimes hypergranularity is seen (Fig. 20.2B). The leukocyte alkaline phosphatase (LAP) score may be low.30 Rarely, giant granules typical of those seen in Chediak–Higashi syndrome have been identified. Auer rods are sometimes seen and, when present, should lead to classification of the disease as RAEB-2, due to their reported negative prognostic significance.31 Accurate enumeration of blast cells in blood has been stressed in WHO 2008. Cases with 2–4% blast cells in blood are classified as RAEB-1, even if the fraction in BM smears is <5%.4 Cases with 1% blast cells in blood and <5% in BM are MDS-unclassified.
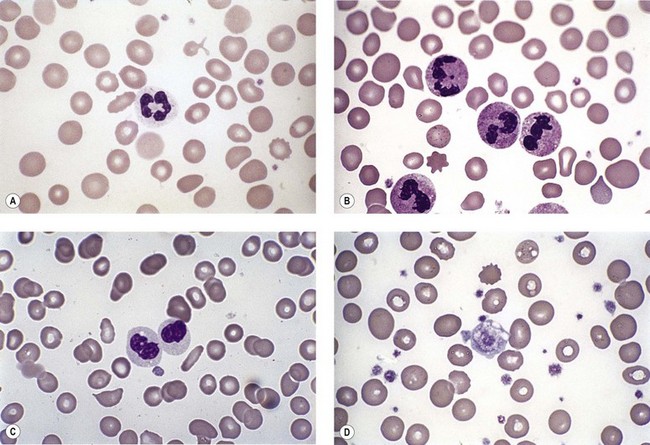
Abnormal neutrophil nuclear lobation is the other feature to observe. Pseudo-Pelger cells are neutrophils with unilobated or bilobed nuclei with normal condensation of the nuclear chromatin (Fig. 20.2C). It is important to distinguish these cells from myelocytes and metamyelocytes, in which the chromatin remains relatively uncondensed. Frequently, pseudo-Pelger cells display hypogranular cytoplasm and, occasionally, neutrophils with hyperlobated nuclei are seen. An unusual feature is arachnoid lobation, with the nuclear lobes looking like the segments of a spider’s leg.
A relative increase of monocytes can be seen in MDS due to neutropenia while chronic myelomonocytic leukemia (currently classified by the WHO as a myelodysplastic/myeloproliferative neoplasm, see Chapter 27) is a differential diagnosis in patients with a total monocytosis >1 × 109/l. Degranulated eosinophils are sometimes a feature of MDS. Eosinophils with basophilic granules, or vice versa, are sometimes seen. Significant eosinophilia and/or basophilia is seen in approximately 12% of MDS patients and predicts poorer prognosis.32
Platelets
Thrombocytopenia is common. Giant platelets or megakaryocyte fragments may circulate in MDS (Fig. 20.2D) and agranular platelets may also be seen.
Thrombocytosis is rare, but occurs in MDS with del(5q). It is important to distinguish myeloproliferative thrombocythemia from the thrombocytosis of MDS. The former should not have dysplastic features, although anemia with ring sideroblasts has been described accompanying marked thrombocytosis, histological BM features resembling essential thrombocythemia and (in most cases) presence of JAK2V617F mutation (see Chapter 27).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


