Myelodysplastic Syndromes
KEY CONCEPTS
![]() Myelodysplastic syndromes (MDS) primarily affect elderly adults, with median age at diagnosis of 76 years.
Myelodysplastic syndromes (MDS) primarily affect elderly adults, with median age at diagnosis of 76 years.
![]() MDS are associated with environmental, occupational, and therapeutic exposures to chemicals or radiation.
MDS are associated with environmental, occupational, and therapeutic exposures to chemicals or radiation.
![]() The clonal population of cells manifested as MDS results from enhanced self-renewal of a hematopoietic stem cell or acquisition of self-renewal in a progenitor cell, increased proliferative capacity in the abnormal clone, impaired cell differentiation, evasion of immune regulation, and antiapoptotic mechanisms in the disease-sustaining cell.
The clonal population of cells manifested as MDS results from enhanced self-renewal of a hematopoietic stem cell or acquisition of self-renewal in a progenitor cell, increased proliferative capacity in the abnormal clone, impaired cell differentiation, evasion of immune regulation, and antiapoptotic mechanisms in the disease-sustaining cell.
![]() Most patients with MDS present with fatigue and lethargy or symptoms related to anemia-induced tissue hypoxia.
Most patients with MDS present with fatigue and lethargy or symptoms related to anemia-induced tissue hypoxia.
![]() The prognosis of patients with MDS is variable. Overall survival time ranges from a few months to several years and can be estimated with the International Prognostic Scoring System (IPSS) or International Prognostic Scoring System—Revised (IPSS-R).
The prognosis of patients with MDS is variable. Overall survival time ranges from a few months to several years and can be estimated with the International Prognostic Scoring System (IPSS) or International Prognostic Scoring System—Revised (IPSS-R).
![]() The primary goal of therapy is hematologic improvement for low-risk patients and alteration in the natural course of the disease for high-risk patients. Palliation of symptoms and improvement in quality of life are goals of therapy for all patients.
The primary goal of therapy is hematologic improvement for low-risk patients and alteration in the natural course of the disease for high-risk patients. Palliation of symptoms and improvement in quality of life are goals of therapy for all patients.
![]() Current guidelines recommend erythropoietin (EPO) or darbepoetin for management of anemia in patients with MDS.
Current guidelines recommend erythropoietin (EPO) or darbepoetin for management of anemia in patients with MDS.
![]() Hypomethylating agents are appropriate for patients with low-risk and intermediate-1-risk MDS with clinically significant neutropenia or thrombocytopenia, patients with anemia who are unlikely to respond to or have not responded to a trial of EPO, and patients who qualified for and failed immunosuppressive therapy.
Hypomethylating agents are appropriate for patients with low-risk and intermediate-1-risk MDS with clinically significant neutropenia or thrombocytopenia, patients with anemia who are unlikely to respond to or have not responded to a trial of EPO, and patients who qualified for and failed immunosuppressive therapy.
![]() Antithymocyte globulin is appropriate treatment for low or intermediate-1 IPSS risk, human leukocyte antigen DR15 positive expressing MDS in patients with symptomatic anemia that is unlikely to respond to erythropoietic agents.
Antithymocyte globulin is appropriate treatment for low or intermediate-1 IPSS risk, human leukocyte antigen DR15 positive expressing MDS in patients with symptomatic anemia that is unlikely to respond to erythropoietic agents.
![]() Lenalidomide is the recommended initial treatment for low-risk 5q- syndrome accompanied by symptomatic anemia.
Lenalidomide is the recommended initial treatment for low-risk 5q- syndrome accompanied by symptomatic anemia.
![]() Allogeneic hematopoietic stem cell transplantation offers potentially curative therapy to patients with MDS who have a donor and are healthy enough for the procedure.
Allogeneic hematopoietic stem cell transplantation offers potentially curative therapy to patients with MDS who have a donor and are healthy enough for the procedure.
INTRODUCTION
Myelodysplastic syndromes (MDS) are clonal, heterogenous, stem cell disorders characterized by predominantly hypercellular bone marrows, anemia, thrombocytopenia, leukopenia, and an inherent predisposition toward evolution to acute myeloid leukemia (AML).1–3 The diagnostic hallmark for MDS is the presence of bone marrow dysplasia in at least 10% of cells of a single myeloid lineage.3 The clinical course of patients with MDS varies from a slowly progressing indolent disease to more aggressive disease characterized by excess bone marrow blasts and rapid progression to AML.4
Our understanding of MDS and the available treatment options have advanced in recent years. Based on new scientific and clinical information, the World Health Organization (WHO) revised its classification system in 2008 to refine diagnostic criteria and the subcategorization of MDS.1 Genetic aberrations in hematopoietic transcription factors, methylation regulators, spliceosome components, and tumor suppressor genes have redefined the molecular landscape in MDS and have been incorporated into prognostic models predicting survival and leukemic transformation.5–7 Between 2004 and 2006, three medications (azacitidine, decitabine, and lenalidomide) were approved by the FDA for the treatment of MDS. In 2009, the first randomized controlled clinical trial that showed a survival advantage for a therapeutic intervention in MDS was published, and several more randomized trials have been published or are ongoing.8 The change in classification of MDS, improvement in risk stratification, and development of new treatment options represent important steps forward in MDS.
EPIDEMIOLOGY
![]() MDS primarily affect elderly adults, with a median age at diagnosis of 76 years and a male predominance, with an estimated male-to-female ratio of about 1.75 to 1.9,10 Overall, an estimated 3 to 12 cases of MDS are diagnosed per 100,000 persons per year. The incidence of MDS increases with age; in patients older than 65 to 70 years, an estimated 27 to 75 new cases occur per 100,000 persons per year.11,12 The Surveillance, Epidemiology and End Results (SEER) Program estimates about 19,600 new cases of MDS are diagnosed in the United States each year.10 Recent reports suggest that the incidence of MDS has been grossly underestimated, with an analysis of a Medicare claims database indicating it could be as high as 45,000 per year.12 Many experts predict that the incidence of MDS will increase as the population of the United States ages and clinicians become more aware of MDS.9
MDS primarily affect elderly adults, with a median age at diagnosis of 76 years and a male predominance, with an estimated male-to-female ratio of about 1.75 to 1.9,10 Overall, an estimated 3 to 12 cases of MDS are diagnosed per 100,000 persons per year. The incidence of MDS increases with age; in patients older than 65 to 70 years, an estimated 27 to 75 new cases occur per 100,000 persons per year.11,12 The Surveillance, Epidemiology and End Results (SEER) Program estimates about 19,600 new cases of MDS are diagnosed in the United States each year.10 Recent reports suggest that the incidence of MDS has been grossly underestimated, with an analysis of a Medicare claims database indicating it could be as high as 45,000 per year.12 Many experts predict that the incidence of MDS will increase as the population of the United States ages and clinicians become more aware of MDS.9
ETIOLOGY
![]() The exact cause of MDS is unknown and is probably multifactorial. MDS have been associated with environmental, occupational, and therapeutic exposures to chemicals or radiation.13 Environmental exposure to agricultural chemicals has been associated with an increased risk of developing MDS.13,14 MDS have also been linked in a dose-dependent relationship to ionizing radiation in atomic bomb survivors in Japan and have been reported in workers in the Chernobyl nuclear accident.15 Occupational exposures to hair dyes, cereal dusts, exhaust gases, diesel fuel, and industrial solvents (including benzene and toluene) have been associated with development of MDS.13,14 Individuals with a family history of a hematologic malignancy are at increased risk for developing MDS.13,14 Modifiable risk factors for MDS include smoking and obesity.16 Recent data suggest that chronic immune stimulation or therapy to manage infectious and autoimmune diseases increases the risk for development of MDS.17
The exact cause of MDS is unknown and is probably multifactorial. MDS have been associated with environmental, occupational, and therapeutic exposures to chemicals or radiation.13 Environmental exposure to agricultural chemicals has been associated with an increased risk of developing MDS.13,14 MDS have also been linked in a dose-dependent relationship to ionizing radiation in atomic bomb survivors in Japan and have been reported in workers in the Chernobyl nuclear accident.15 Occupational exposures to hair dyes, cereal dusts, exhaust gases, diesel fuel, and industrial solvents (including benzene and toluene) have been associated with development of MDS.13,14 Individuals with a family history of a hematologic malignancy are at increased risk for developing MDS.13,14 Modifiable risk factors for MDS include smoking and obesity.16 Recent data suggest that chronic immune stimulation or therapy to manage infectious and autoimmune diseases increases the risk for development of MDS.17
About 10% to 15% of all cases of MDS are attributed to radiation, chemotherapy, or both and are termed therapy-related MDS (t-MDS).18–20 More aggressive chemotherapy regimens and improved survival after cancer treatment are contributing to an increased incidence of t-MDS.20,21 t-MDS have an increased likelihood of progression to AML and a poorer prognosis than de novo MDS.19,20,22 Chromosomal abnormalities are found in about 90% of t-MDS compared with 50% to 60% of de novo MDS.23–25
The risk for developing t-MDS increases with age, higher doses of chemotherapy or radiation, longer duration of exposure, and exposure to both chemotherapy and radiation.14,22,23 Several chemotherapeutic agents have been associated with t-MDS (Table 114-1). The contribution of a specific agent is difficult to assess because patients are usually exposed to multiple agents, often in combination with radiation.22 The most frequently reported classes of chemotherapeutic agents associated with t-MDS are alkylating agents and topoisomerase II inhibitors.18,19,23,24
TABLE 114-1 Therapies Associated with Therapy-Related Myelodysplastic Syndrome
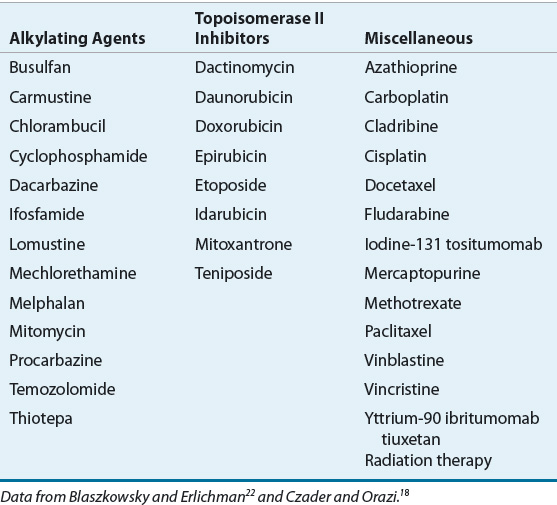
The role of alkylating agents in the development of t-MDS is well established in patients with cancer and those receiving high cumulative doses of alkylating agents for autoimmune disorders such as rheumatoid arthritis.14,18,24 The latency period between exposure to alkylating agents and the development of t-MDS is about 4 to 7 years. Characteristic chromosomal abnormalities in t-MDS associated with alkylating agents include deletions on chromosome 5 and chromosome 7.18
Topoisomerase II inhibitors, including the epipodophyllotoxins (etoposide and teniposide), anthracyclines (daunorubicin, doxorubicin, epirubicin, idarubicin), and the anthracenedione mitoxantrone, are also associated with t-MDS. t-MDS associated with topoisomerase II inhibitors typically occurs a median of 2 to 3 years after exposure, and patients are more likely to present with AML at diagnosis.14 Chromosomal abnormalities often found in patients with t-MDS associated with topoisomerase II inhibitors include balanced translocations involving the MLL gene 11q23 and 21q22.18
Radioimmunoconjugates, including ibritumomab tiuxetan and iodine-131 tositumomab, are monoclonal antibodies linked to radioactive isotopes. Radiation is delivered to the antibody-bound targeted cell and to neighboring cells through a “cross-fire” effect. t-MDS or AML is reported to occur in 5% to 10% of patients exposed to iodine-131 tositumomab and in 1% to 5% of patients exposed to ibritumomab tiuxetan.26–30 About 8% of patients receiving myeloablative doses of ibritumomab tiuxetan as conditioning regimen before hematopoietic stem cell transplantation (HSCT) developed t-MDS or AML, similar to the rate in patients receiving myeloablative chemotherapy-based conditioning regimens.31 Both agents are used to treat non-Hodgkin lymphoma, a patient population likely to receive other therapies associated with t-MDS, including alkylating agents, anthracyclines, and radiation. Therefore, it is difficult to determine the additional risk for t-MDS due solely to exposure to one of these agents.26,28,30
Granulocyte colony-stimulating factor (G-CSF) use during treatment of solid tumors and lymphoma in adults has been associated with an increased risk for t-MDS.32 A systematic review of 25 randomized controlled trials comparing patients receiving filgrastim or lenograstim with placebo found an absolute risk increase of 0.41% for development of t-MDS or AML associated with use of a colony-stimulating factor.32 The absolute risk of death was 3.4% lower in the group of patients randomized to a hematopoietic growth factor; the benefit was attributed to lower cancer-related mortality because of a greater chemotherapy dose intensity. This systematic review indicates the administration of hematopoietic growth factors to prevent complications associated with febrile neutropenia outweighs the increased t-MDS risk.21 The risk of development of t-MDS may also be higher in patients with congenital neutropenia and aplastic anemia treated with long-term G-CSF.18,33,34
Patients undergoing autologous HSCT are at increased risk for development of t-MDS. Conditioning regimens given before HSCT usually include high doses of alkylating agents or etoposide, often in combination with total-body irradiation. As many as 8% to 20% of patients with non-Hodgkin lymphoma treated with autologous HSCT will be diagnosed with t-MDS within 10 years of transplantation.35–37 Risk factors for development of t-MDS after autologous HSCT include antecedent conventional chemotherapy, prior radiation therapy, low stem cell dose, older age at time of transplant, and use of total-body irradiation in the conditioning regimen.35,36,38
PATHOPHYSIOLOGY
![]() Knowledge of normal hematopoiesis is needed to understand the pathophysiology of MDS (see eChap. 20 for a more detailed description of hematopoiesis). Progressive bone marrow failure is characteristic of patients with MDS and is the result of ineffective hematopoiesis. In addition to peripheral blood cytopenias, the terminally differentiated cells that are produced may have functional defects. Neutrophils may have reduced bactericidal and fungicidal activity despite a normal quantity of neutrophils.39 Platelets may be normal in quantity but have impaired activation, secretion, and aggregation.40 The diverse pathophysiology underlying MDS causes the heterogeneity in clinical presentation, pattern of disease progression, and response to therapy and has not been fully elucidated.41 A multistep model for the pathogenesis of MDS has been proposed, and it is likely that the disease can arise via multiple different pathways.3,13,41 The clonal population of cells manifested as MDS results from enhanced self-renewal of a hematopoietic stem cell or acquisition of self-renewal in a progenitor cell, increased proliferative capacity in the abnormal clone, impaired cell differentiation, evasion of immune regulation, and antiapoptotic mechanisms in the disease-sustaining cell.41 The abnormal clone proliferates or evades apoptosis because of genomic instability and abnormalities in cytokines and the bone marrow stroma.3,41 These changes create a dysplastic, clonal population of cells in a milieu unable to support normal hematopoiesis.
Knowledge of normal hematopoiesis is needed to understand the pathophysiology of MDS (see eChap. 20 for a more detailed description of hematopoiesis). Progressive bone marrow failure is characteristic of patients with MDS and is the result of ineffective hematopoiesis. In addition to peripheral blood cytopenias, the terminally differentiated cells that are produced may have functional defects. Neutrophils may have reduced bactericidal and fungicidal activity despite a normal quantity of neutrophils.39 Platelets may be normal in quantity but have impaired activation, secretion, and aggregation.40 The diverse pathophysiology underlying MDS causes the heterogeneity in clinical presentation, pattern of disease progression, and response to therapy and has not been fully elucidated.41 A multistep model for the pathogenesis of MDS has been proposed, and it is likely that the disease can arise via multiple different pathways.3,13,41 The clonal population of cells manifested as MDS results from enhanced self-renewal of a hematopoietic stem cell or acquisition of self-renewal in a progenitor cell, increased proliferative capacity in the abnormal clone, impaired cell differentiation, evasion of immune regulation, and antiapoptotic mechanisms in the disease-sustaining cell.41 The abnormal clone proliferates or evades apoptosis because of genomic instability and abnormalities in cytokines and the bone marrow stroma.3,41 These changes create a dysplastic, clonal population of cells in a milieu unable to support normal hematopoiesis.
Bone Marrow Microenvironment
The myelodysplastic clone is associated with cellular dysfunction, including excess secretion of cytokines, defective differentiation, genomic instability, and reduced response to regulatory cytokines.3 In contrast to the peripheral blood cytopenias characteristic of MDS, bone marrow cells often have a paradoxically high rate of cellular division. Apoptosis, or programmed cell death, also is increased, leading to futile cycling of precursor cells and impaired production of mature peripheral blood cells.13 Overproduction of proapoptotic and inflammatory cytokines and vascular endothelial growth factor may contribute to this process.41 Bone marrow stromal cells from MDS patients show decreased ability to support normal hematopoietic cell function.13
Patients with MDS frequently have evidence of immune dysregulation, such as impaired immune surveillance and autoimmune reactions.3 Cytopenias can be related to an autoimmune T-cell–mediated response. A subset of MDS patients characterized by younger age, refractory anemia of short duration, a hypocellular bone marrow, trisomy 8 as the sole cytogenetic abnormality, and expression of human leukocyte antigen (HLA) haplotype DR15 have a high likelihood of response to immunosuppressive therapy.13,41 Immunosuppressive therapy with cyclosporine and antithymocyte globulin may induce durable responses in this subgroup of patients, confirming the role of immune dysregulation.3 Whether B cells and T cells are a part of the MDS clonal population or a secondary reaction after the development of the malignant clone is unclear.13
Genomic Instability
In the multistep model for development of MDS, one or more transformations occur that confer a growth advantage to the dysplastic cell, leading to the emergence of a clonal population.41 The inciting transformations in genetic material have not been identified. Chromosomal abnormalities, most often genomic losses and gains, are detected by cytogenetic analysis in about 50% of patients with de novo MDS and remain one of the strongest determinants of prognosis.4,20,25,42–44 Multiple cytogenetic abnormalities that correlate with the clinical course of MDS were incorporated in the original International Prognostic Scoring System (IPSS) classification and prognostic assessment, including 5q or 20q deletions and chromosome 7 abnormalities.4 The newly revised IPSS classification (International Prognostic Scoring System–Revised [IPSS-R]) includes several additional cytogenetic abnormalities, including trisomy 8 or 19, 12p or 11q deletions, and double abnormalities, that correlate with the clinical course of MDS.7 Deletions on chromosome 5q occur in up to 12% of patients and are of particular interest because multiple genes involved in hematopoiesis are located there.25 Additionally, MDS with 5q deletions as the sole genetic aberration are recognized as a distinct subtype of MDS with a favorable prognosis and a high likelihood of response to lenalidomide.25,45
High-resolution single nucleotide polymorphism array, a new type of karyotypic analysis that can detect molecular abnormalities that are too minute for traditional metaphase cytogenetics, has shown independent prognostic value for MDS.6,46 One of the most frequently occurring molecular alterations, seen in 22% to 26% of MDS patients, is alteration of TET2 protein thought to be responsible for passive DNA methylation. Mutation in TET2 has been identified as a favorable prognostic factor, although results were not confirmed in larger confirmatory studies.47–49 Transformations in additional tumor suppressor genes, transcription factors, and cell cycle regulators such as ASXL1, RUNX1, EZH2, DNMT3A, and several others are also thought to provide the dysplastic stem cell with a growth advantage and offer additional prognostic insight.50
Epigenetics
In addition to changes detected on chromosomal analysis, several transformations have been identified that may contribute to myelodysplasia that do not result from alteration of the nucleic acid sequence in DNA. The term epigenetics refers to mechanisms that regulate the expression of DNA without affecting its sequence. Epigenetic changes have been identified in numerous malignancies and are of particular importance in the context of MDS.51
DNA methylation is the best described and most common epigenetic marker. In the mammalian genome, only cytosine located 5′ to a guanosine (CpG) can be methylated (CpG pair). Clusters of CpG pairs, known as CpG islands, are near the promoter regions for many genes. These regions are unmethylated in normal cells, allowing for standard DNA expression to occur.51 Increased methylation (hypermethylation) of CpG islands occurs via DNA methyltransferase and is associated with aberrant gene silencing, which may lead to further genetic instability and dysfunction of the cell cycle. Decreased methylation (hypomethylation) may lead to reexpression of previously silenced genes.51 Hypermethylation and gene silencing have been identified in patients with MDS, and azacitidine and decitabine reverse this process.51,52
Histone acetylation, a second epigenetic marker, is also gaining significance in MDS.53 Histones coil with DNA to form tightly wound complexes called chromatin. Posttranslational modifications of histones, by acetylation or deacetylation, can alter the structure of chromatin creating opportunities for gene suppression or expression, depending on the structural change of the chromatin.53 Histone hypoacetylation has been documented in malignant cells and several histone deacetylase inhibitors have been studied in patients with MDS as monotherapy or in combination with hypomethylating agents in an attempt to promote histone acetylation and expression of previously suppressed tumor suppressor genes.54,55
CLASSIFICATION AND PROGNOSIS
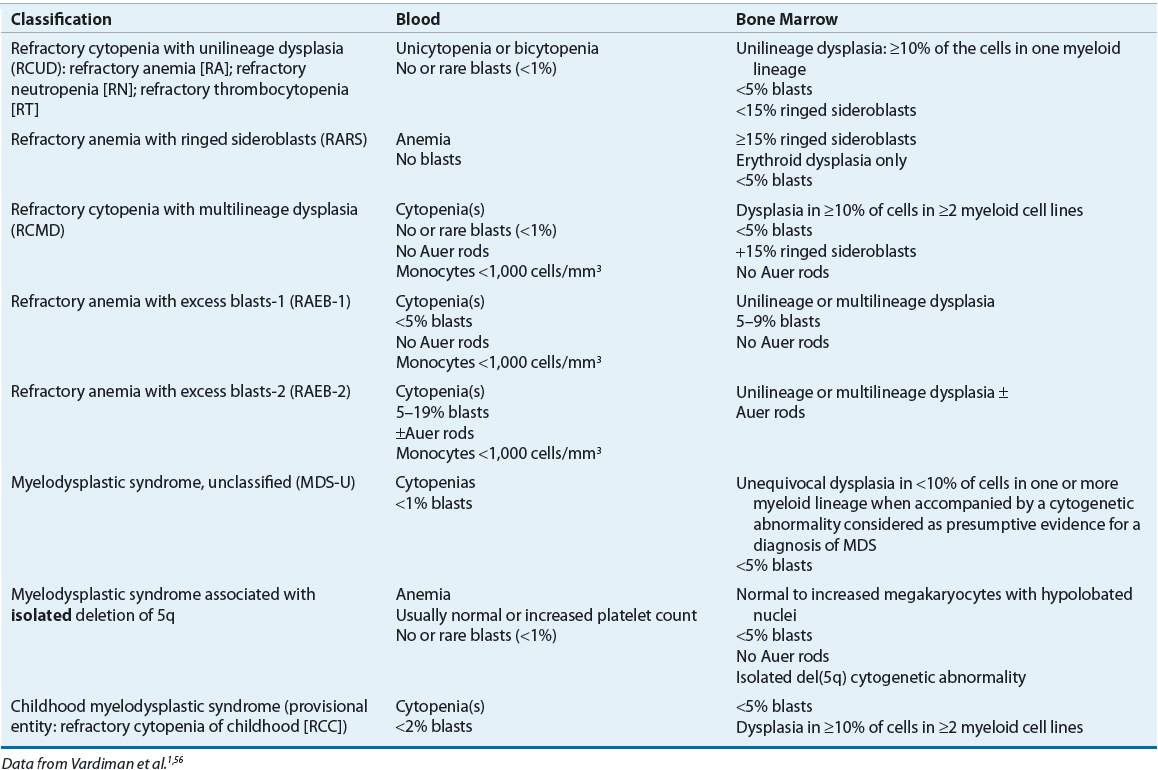
Several classification systems and models for predicting risk in MDS have been developed. The French-American-British (FAB) and WHO are classification schemes for MDS. The FAB classification established subgroups of MDS based on morphology of bone marrow aspirates and peripheral blood blast percentage.14 In the 2008 WHO classification system, MDS are categorized based on bone marrow and peripheral blood blast percentage, cytogenetics, and whether dysplasia or cytopenias affect a single cell lineage or multiple myeloid cell lines (Table 114-2).56 The most significant changes in the WHO classification compared with the FAB classification include the following: patients with a single cell lineage affected are separated from those with multiple myeloid cell lines affected, patients with greater than 20% blasts in the marrow are now considered to have acute leukemia, patients with deletion on chromosome 5 are now in a distinct category, and chronic myelomonocytic leukemia is classified as a myelodysplastic/myeloproliferative disorder. Two studies showed the ability of the WHO classification to identify patient subgroups with differences in survival and responses to erythropoietin (EPO) and filgrastim. Patients with refractory anemia or refractory anemia with ringed sideroblasts (RARS) had prolonged overall and leukemia-free survival and improved response to therapy compared with those with refractory cytopenia with multilineage dysplasia or refractory anemia with excessive blasts.57 This classification scheme may help predict the prognosis of MDS, but it has not been shown to predict response to a given therapy.
TABLE 114-2 World Health Organization Classification of Myelodysplastic Syndromes
CLINICAL PRESENTATION AND DIAGNOSIS Myelodysplastic Syndromes
General
• ![]() Patients with MDS may develop isolated anemia (hemoglobin <11 g/dL), neutropenia (<1,500 cells/mm3), or thrombocytopenia (<100 × 109/L) or multiple peripheral cytopenias.
Patients with MDS may develop isolated anemia (hemoglobin <11 g/dL), neutropenia (<1,500 cells/mm3), or thrombocytopenia (<100 × 109/L) or multiple peripheral cytopenias.
• Patients may be asymptomatic, with cytopenia(s) discovered on complete blood count with differential.
Symptoms
• If symptomatic, the patient may report fatigue, lethargy, malaise, palpitations, dyspnea on exertion, or other symptoms associated with hypoxia secondary to anemia.
• Patients may have symptoms of infection, including cough or dysuria.
• Patients may present with complaints of easy bruising or bleeding.
Signs
• Pallor, tachycardia, or tachypnea related to anemia
• Fever, chills, rigors caused by infection and immune dysfunction
• Petechiae, bruising, epistaxis, gingival bleeding, excessive vaginal bleeding, bruising, or hematuria caused by thrombocytopenia
Laboratory Tests
• Complete blood count with differential
• Anemia often is macrocytic or normocytic with a low reticulocyte index
• Serum vitamin B12, red blood cell (RBC) folate and copper levels
• Testing for the human immunodeficiency virus (HIV)
• Serum thyroid-stimulating hormone
• Serum EPO level
• Serum ferritin, iron, and total iron-binding capacity
Other Diagnostic Tests
• Bone marrow biopsy and aspirate: send for morphologic examination, cytochemical staining, immunophenotyping, and cytogenetics (chromosome analysis).
• Repeat bone marrow biopsy of patients with MDS often is required because dysplastic features may progress over time or the bone marrow may be unevenly distributed.
Criteria for Diagnosis
• Stable cytopenia for at least 6 months (2 months if accompanied by a specific karyotype associated with MDS or bilineage dysplasia)
• Exclusion of other causes of cytopenia or dysplasia
• One of the following:
1. Dysplasia (>10% in one or more of three major bone marrow lineages)
2. Blast cell count of 5% to 19%
3. Specific MDS-associated karyotype (e.g., del(5q), del(20q), +8, or del(7q))
Data from Tefferi and Vardiman,3 Faderl and Kantarjian,14Valent et al.,161and The NCCN Clinical Practice Guidelines in Oncology™.61
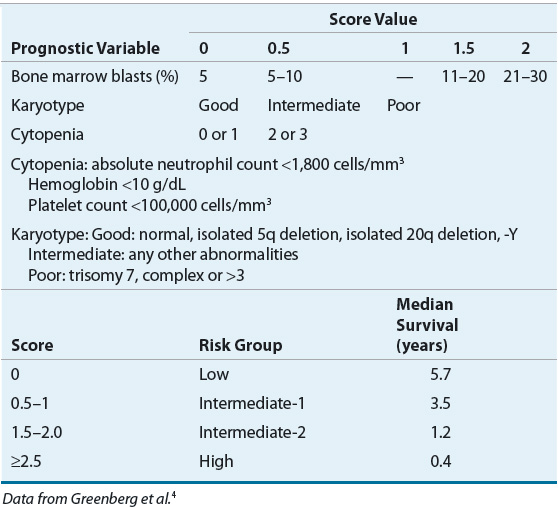
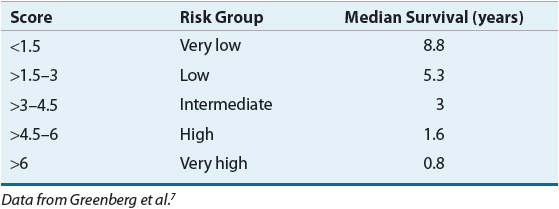
Models to predict overall survival and risk of transformation to AML continue to be developed and refined as new information about the genetic basis of MDS evolves. The IPSS is still used in clinical trials and continues to guide therapy as newer models are developed and validated.3,5 ![]() Based on an observational study of mostly untreated MDS patients, the IPSS was developed to identify factors that would predict progression of MDS.4 Multivariate analyses identified four prognostic factors: cytogenetic abnormalities, percentage of bone marrow blasts, age, and number of cytopenias. Using these four factors, researchers were able to stratify patients into four risk groups that correlated with overall survival, which ranged from a few months to several years (Table 114-3).
Based on an observational study of mostly untreated MDS patients, the IPSS was developed to identify factors that would predict progression of MDS.4 Multivariate analyses identified four prognostic factors: cytogenetic abnormalities, percentage of bone marrow blasts, age, and number of cytopenias. Using these four factors, researchers were able to stratify patients into four risk groups that correlated with overall survival, which ranged from a few months to several years (Table 114-3).
TABLE 114-3 International Prognostic Scoring System for Myelodysplastic Syndromes
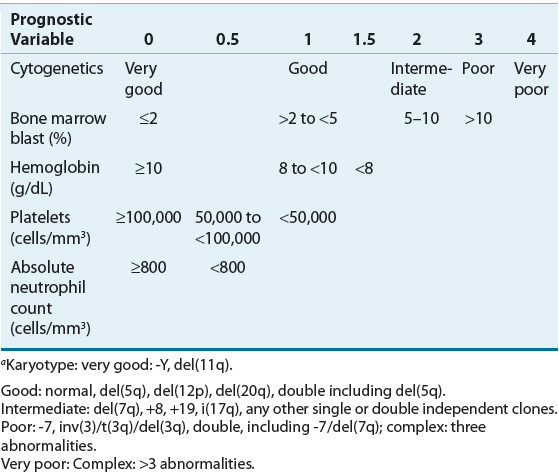
The IPSS-R was developed after analysis of more than 7,000 patients whose disease had not been treated with disease-altering therapy (Table 114-4).7 This model differs from the IPSS by identifying five risk categories by incorporating different categories for marrow blast percentage value and depth of cytopenias, expanding the cytogenetic risk groups from three to five groups and including a number of less common cytogenetic abnormalities. Patient age, performance status, and serum ferritin and lactate dehydrogenase levels were additional significant predictors for survival but not for AML transformation. Recent reports have demonstrated that recurrent somatic gene mutations predict prognosis independent of the IPSS score, and their incorporation into a risk classification scheme does enhance the model.6,58 About half of patients with MDS have normal cytogenetics; therefore, after the full spectrum of somatic mutations in MDS has been defined, optimal prognostic scoring systems will need to include relevant molecular features. No single method of predicting risk of disease progression, overall survival, or response to therapy has been universally adopted.5 The risk prognostic schemas are derived from patients with primary, untreated MDS and may not be applicable to patients who have t-MDS, those who received disease-modifying therapy, or those who have undergone karyotypic evolution in the course of their disease.
TABLE 114-4 International Prognostic Scoring System—Revised for Myelodysplastic Syndromesa
Clinical Controversy…
Currently, the FAB classification is used for coding and billing purposes, for drug indication languages approved by regulatory agencies, and for some clinical trial inclusion or evaluation criteria.5 The WHO criteria are used by pathologists to describe MDS and in both clinical trial and off-study management of patients with MDS. The IPSS is the most widely used prognostic scoring system for MDS patients enrolling in clinical trials and is used to guide therapy in the United States.3,5 The IPSS-R will likely be adopted by most centers until the role of molecular information in the prognosis of MDS is better defined.59
TREATMENT
Treatment of MDS has rapidly evolved during the last decade following discoveries in disease biology, introduction of new methods for predicting the natural history of the disease and response to a given therapy, and development of new treatment strategies (Fig. 114-1).
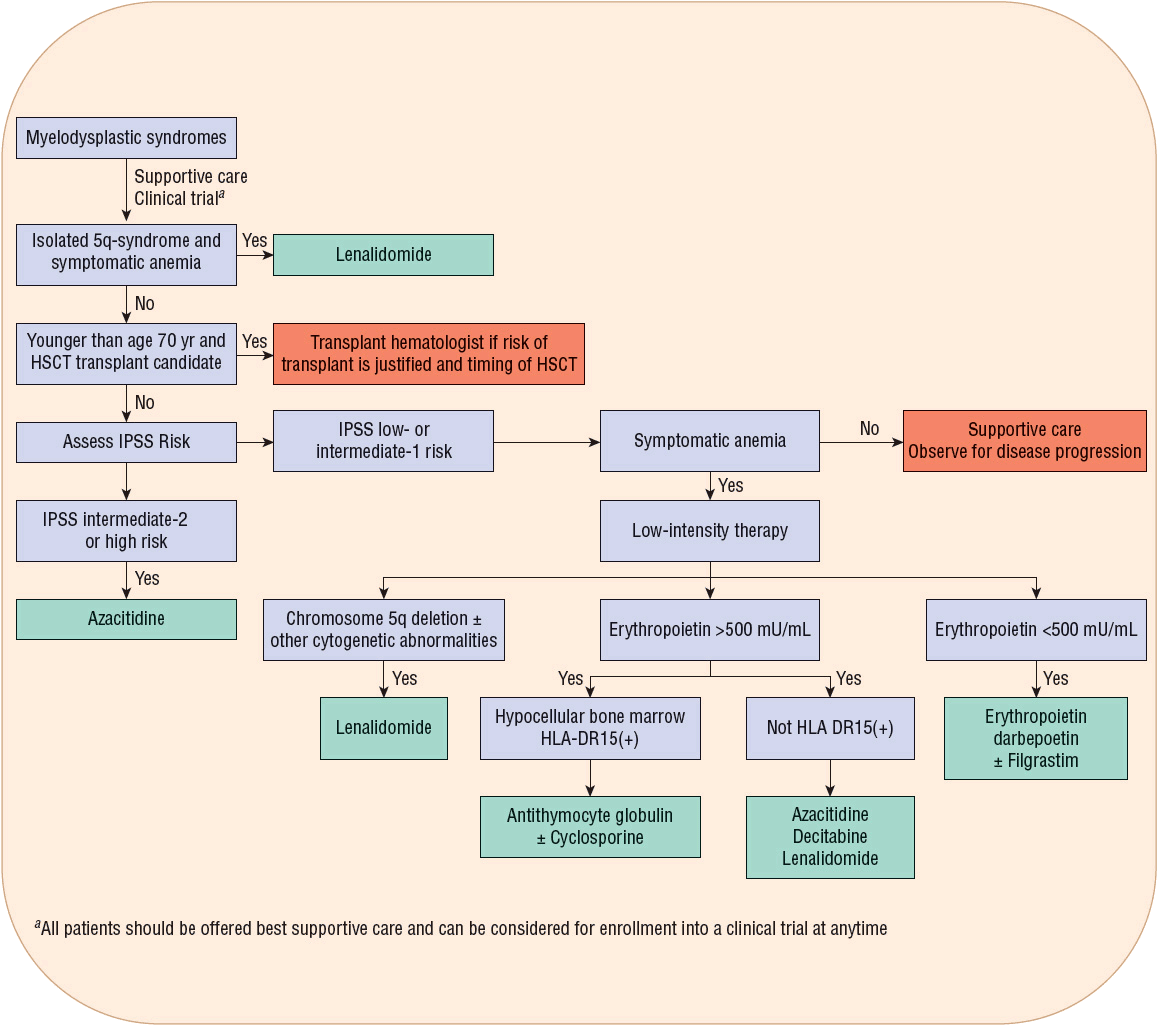
FIGURE 114-1 Myelodysplastic syndromes treatment algorithm. (HSCT, hematopoietic stem cell transplant; IPSS, International Prognostic Scoring System.)
Desired Outcomes
![]() The goals of treatment vary with disease-specific factors, including the type of MDS; cytogenetics; risk of progression to AML and death; rate of disease progression; and patient factors, including age, organ function, performance status, and presence of symptoms related to myelodysplasia.60 The primary goal of therapy is hematologic improvement for low-risk patients and alteration in the natural course of the disease for high-risk patients.61 Additional critical goals for all risk groups include symptom palliation and quality-of-life improvement. Lower-intensity treatment with a DNA hypomethylating agent or immunosuppressive therapy may improve overall survival, provide symptom palliation, and enhance quality of life without significant toxicity.8,60,61 The only curative therapy for MDS is allogeneic HSCT, but most patients lack a suitable donor, are not healthy enough to undergo this intensive therapy, or may not be referred for HSCT because of advanced biologic age despite adequate health and organ function.3,62
The goals of treatment vary with disease-specific factors, including the type of MDS; cytogenetics; risk of progression to AML and death; rate of disease progression; and patient factors, including age, organ function, performance status, and presence of symptoms related to myelodysplasia.60 The primary goal of therapy is hematologic improvement for low-risk patients and alteration in the natural course of the disease for high-risk patients.61 Additional critical goals for all risk groups include symptom palliation and quality-of-life improvement. Lower-intensity treatment with a DNA hypomethylating agent or immunosuppressive therapy may improve overall survival, provide symptom palliation, and enhance quality of life without significant toxicity.8,60,61 The only curative therapy for MDS is allogeneic HSCT, but most patients lack a suitable donor, are not healthy enough to undergo this intensive therapy, or may not be referred for HSCT because of advanced biologic age despite adequate health and organ function.3,62
General Approach
Therapy for MDS is determined by symptoms, IPSS risk for progression to AML or death, patient age and comorbidities, likelihood of response to a given therapy and its effects on quality of life, and patients’ treatment preferences.2 Patients with extensive, life-limiting comorbidities or who are asymptomatic at diagnosis may warrant supportive care alone.2 About 30% to 60% of patients with MDS receive supportive care alone.25,63 Lower-risk patients are thought to have better prognoses, and thus less toxic therapies are used to manage MDS, including EPO, darbepoetin, lenalidomide, or DNA hypomethylating agents. Patients with intermediate-2 and high IPSS risk MDS have poorer prognoses and may be candidates for allogeneic HSCT; patients who are not HSCT candidates may benefit from a DNA hypomethylating agent.8,61 As outcomes of therapeutic interventions for all risk categories are identified and genetic markers increase the ability of clinicians to stratify patients into even more specific risk categories, the line between therapies for high- and low-risk patients has been blurred.20 Clinicians should recognize that the clinical course of MDS is not static. MDS may progress; comorbidities or symptoms may change over time along with the ability to create more personalized medicine, necessitating an adjustment in treatment strategy. Therapy for MDS is generally palliative, and enrollment in a suitable clinical trial is always a viable approach.2,64
Careful interpretation is necessary when comparing the results of clinical trials in MDS because patient characteristics and response criteria vary widely. Described previously, the clinical course and prognosis are affected by patient-specific characteristics.4,65 Examples of different response criteria used include changes in hemoglobin, changes in RBC transfusion requirements, or effects on quality of life.60 The use of RBC transfusion requirement as a primary end point is especially problematic because decisions concerning RBC transfusion needs are highly individualized and may not be consistent among clinicians. Additionally, the relationship between changes in hemoglobin or decreases in RBC transfusion requirements and improved quality of life is not clear. Some treatments for MDS can cause significant adverse effects, resulting in hospitalization or increased clinic visits, and may negatively impact quality of life regardless of their positive effects on hematologic parameters. The impact of treatment on quality of life is an important consideration when selecting therapy and should be assessed regularly with the use of validated instruments.
Supportive Care
All patients with MDS should receive supportive care, including clinical monitoring, psychosocial support, and quality-of-life assessment.61 The National Comprehensive Cancer Network (NCCN) guidelines recommend that patients with symptomatic anemia should receive leukoreduced RBC transfusions, and those with bleeding caused by thrombocytopenia or platelet counts below 10,000 cells/mm3 (10 × 109/L) should receive platelet transfusions.61 Hematopoietic growth factor support should be considered in patients with refractory, symptomatic cytopenias. Patients with evidence of infection should have an appropriate diagnostic evaluation based on history and physical examination followed by appropriate antimicrobial therapy. Routine antimicrobial or hematopoietic growth factor prophylaxis is not recommended in the absence of repeated infections. Iron chelation may be considered in low-risk and intermediate-1-risk patients and candidates for allogeneic HSCT who have received more than 20 to 30 RBC transfusions and are expected to continue to require transfusions.61
Infection
Patients with MDS may be neutropenic or have functional defects in neutrophils, predisposing them to infection.39 In MDS, the most frequently isolated organisms are bacteria, and the most common sites of infection are the lungs, urinary tract, and bloodstream.66 Patients with evidence of infection should have appropriate diagnostic evaluation based on history and physical examination and then appropriate antimicrobial therapy. Neutropenic patients with evidence of infection or fever of unknown origin should receive empiric broad-spectrum, IV antibiotics.67
Hematopoietic Growth Factors
Filgrastim (G-CSF) and sargramostim (granulocyte-macrophage colony-stimulating factor [GM-CSF]) are colony-stimulating factors that stimulate white blood cell production and may increase circulating neutrophils in 70% to 90% of patients, which may decrease risk of infection.68 These agents have not been shown to be beneficial as chronic monotherapy because they do not reliably prevent infection and have no impact on survival.68 G-CSF or GM-CSF should only be administered temporarily as monotherapy in the rare neutropenic MDS patient who develops recurrent severe infections.2,3
EPO is a protein produced by the kidney in response to hypoxia that stimulates proliferation and differentiation of erythroid cells. Anemic patients with MDS may have either a lower than expected endogenous serum EPO level relative to the degree of anemia present or an elevated EPO level. The mechanism of action of recombinant erythropoiesis-stimulating agents (ESAs) in MDS is not clear, but exogenous EPO may stimulate a normal clone of cells that is unresponsive to low endogenous levels of EPO, stimulate a dysplastic clone to differentiate that is less responsive to endogenous EPO, or induce apoptosis. An immunomodulatory effect of EPO, G-CSF, or GM-CSF has been proposed.
![]() Current guidelines recommend use of ESAs for management of anemia in patients with MDS.61,69 Unlike some solid tumors,70 no detrimental effects on overall survival or progression to leukemia have been noted in patients with MDS. Treatment with ESAs alone may result in hematologic improvement and transfusion independence in low- and intermediate-1 IPSS risk patients. Two meta-analyses have evaluated the efficacy of ESAs in MDS. The first analysis, which included 2,106 patients from 59 studies reported between 1990 and 2005, found a hemoglobin response of about 30% based on the definition of hemoglobin response in the original publication.71 A subsequent meta-analysis only included studies from 1990 to 2006 that reported results by International Working Group (IWG) criteria60 to define erythroid response (an increase in hemoglobin of 2 g/dL [1.24 mmol/L] or transfusion independence). This report included 30 studies with 925 patients with MDS and found an overall erythroid response rate of 58% in patients receiving ESAs.72 The latter report also suggests that EPO and darbepoetin can be used interchangeably for the management of MDS based on similar response rates achieved. The higher response rate compared with the previous meta-analysis likely reflects inclusion of a higher proportion of patients most likely to respond to ESAs. Patients with low- and intermediate-1 IPSS risk MDS who have a serum EPO level less than 500 mU/mL (500 IU/L) and a history of receiving fewer than 2 units of RBC transfusions per month have the best chance at responding to ESAs.2,73 The doses required to achieve a response in MDS are higher than those used to treat renal causes of anemia, with EPO doses in the range from 40,000 to 60,000 units subcutaneously two to three times per week.61 Darbepoetin doses ranging from 100 to 300 mcg subcutaneously weekly or every other week have also been used for MDS management.2,72 Doses should be titrated up or down, as clinically indicated, to achieve a hemoglobin level of 10 to 12 g/dL (6.2 to 7.44 mmol/L).74 Additionally, patients should receive at least 8 weeks of therapy before doses are adjusted or before patients are considered nonresponders because response to ESAs in MDS can be delayed.2,61 The median response duration for ESAs in MDS is 1 to 2 years, and the ESA should be discontinued if there is no benefit or the response wanes.2
Current guidelines recommend use of ESAs for management of anemia in patients with MDS.61,69 Unlike some solid tumors,70 no detrimental effects on overall survival or progression to leukemia have been noted in patients with MDS. Treatment with ESAs alone may result in hematologic improvement and transfusion independence in low- and intermediate-1 IPSS risk patients. Two meta-analyses have evaluated the efficacy of ESAs in MDS. The first analysis, which included 2,106 patients from 59 studies reported between 1990 and 2005, found a hemoglobin response of about 30% based on the definition of hemoglobin response in the original publication.71 A subsequent meta-analysis only included studies from 1990 to 2006 that reported results by International Working Group (IWG) criteria60 to define erythroid response (an increase in hemoglobin of 2 g/dL [1.24 mmol/L] or transfusion independence). This report included 30 studies with 925 patients with MDS and found an overall erythroid response rate of 58% in patients receiving ESAs.72 The latter report also suggests that EPO and darbepoetin can be used interchangeably for the management of MDS based on similar response rates achieved. The higher response rate compared with the previous meta-analysis likely reflects inclusion of a higher proportion of patients most likely to respond to ESAs. Patients with low- and intermediate-1 IPSS risk MDS who have a serum EPO level less than 500 mU/mL (500 IU/L) and a history of receiving fewer than 2 units of RBC transfusions per month have the best chance at responding to ESAs.2,73 The doses required to achieve a response in MDS are higher than those used to treat renal causes of anemia, with EPO doses in the range from 40,000 to 60,000 units subcutaneously two to three times per week.61 Darbepoetin doses ranging from 100 to 300 mcg subcutaneously weekly or every other week have also been used for MDS management.2,72 Doses should be titrated up or down, as clinically indicated, to achieve a hemoglobin level of 10 to 12 g/dL (6.2 to 7.44 mmol/L).74 Additionally, patients should receive at least 8 weeks of therapy before doses are adjusted or before patients are considered nonresponders because response to ESAs in MDS can be delayed.2,61 The median response duration for ESAs in MDS is 1 to 2 years, and the ESA should be discontinued if there is no benefit or the response wanes.2
Several trials have evaluated if adding G-CSF to ESAs can enhance the hematologic response, and the conclusions have been inconsistent. Long-term follow-up of 121 patients from three uncontrolled phase II studies was retrospectively compared with that of 237 untreated patients who were matched for FAB classification, hemoglobin, and transfusion needs.75 A 39% major erythroid response rate by IWG criteria was demonstrated in the EPO plus G-CSF cohort. The median doses required to maintain a stable response were subcutaneous EPO 30,000 units/wk and G-CSF 225 mcg/wk.76 In a multivariate analysis, the EPO plus G-CSF group was associated with improved overall survival (hazard ratio [HR], 0.61; 95% confidence interval [CI], 0.44 to 0.83), and decreased risk of nonleukemic death (HR, 0.66; 95% CI, 0.44 to 0.99) compared with the untreated group.75 Retrospective data in 433 patients with MDS treated with EPO or darbepoetin, with or without filgrastim, reported a 50% response rate by IWG 2006 criteria.77 Predictors of response included low- and intermediate-1 IPSS risk, RBC transfusion independence, serum EPO level below 200 mU/mL (200 IU/L), and shorter interval between diagnosis and treatment. The addition of G-CSF was not significantly associated with response. A large phase III, randomized controlled trial of ESAs in MDS with long-term follow-up compared EPO with or without G-CSF to best supportive care in 110 patients.78 At 4 months, 34% of patients receiving EPO had an erythroid response by IWG 2006 criteria compared with 5.8% of patients receiving placebo. A total of 47% of patients had a major erythroid response when EPO doses were escalated or filgrastim was added. Patients with RARS were most likely to respond to the addition of filgrastim. No difference in overall survival or leukemic evolution was observed between patients receiving EPO compared with best supportive care after a median follow-up period of 5.8 years; the study was not prospectively powered to determine differences in these outcomes. To further support the results of this study, a meta-analysis of 15 published trials was performed to compare the erythroid response in patients who received EPO as a single agent with those who received EPO plus G-CSF or GM-CSF.79 The overall erythroid response was 49%, 50.6%, and 64.5% for patients who received standard EPO (30,000 to 40,000 units/wk), standard EPO plus G-CSF or GM-CSF, or high-dose EPO (60,000 to 80,000 units/wk), respectively. The authors concluded that higher doses of single agent EPO are more effective than standard doses alone or in combination with G-CSF or GM-CSF. However, a significantly higher proportion of transfusion-dependent patients were enrolled in the trials using combination therapy compared with the other two treatment groups that could have negatively impacted the outcomes.
Some, but not all, studies have shown that patients who respond to ESAs have improvements in quality of life.78 The value of this costly intervention has not been proven, and long-term safety has not been evaluated in randomized controlled trials.80 Although EPO, with or without G-CSF, does not improve overall survival, it does not shorten overall survival or time to development of leukemia and may decrease the need for RBC transfusions and improve quality of life. ESA therapy is well tolerated, and the NCCN recommends a trial in low- and intermediate-1 IPSS risk patients who have a serum EPO level less than 500 mU/mL (500 IU/L) and a limited transfusion history.61
Thrombopoietin is a hormone synthesized in the liver and secreted into the systemic circulation, where it binds to thrombopoietin receptors on stem cells, progenitor cells, and platelets, resulting in increased platelet production. Romiplostim and eltrombopag are novel drugs that stimulate the thrombopoietin receptor similarly to endogenous thrombopoietin, and are currently FDA approved for patients with chronic idiopathic thrombocytopenic purpura. Two small phase II open-label trials evaluating romiplostim in patients with low- and intermediate-1-risk MDS with platelets less than 50,000 cells/mm3 (50 × 109/L) resulted in durable platelet responses in 30% to 46% of patients by IWG 2000 criteria.81,82 Four of 45 patients developed a transient increase in the proportion of bone marrow blasts, and 2 patients developed AML in one of the studies81; in the other study 2 of 28 patients developed progression to AML.82 A randomized, placebo-controlled trial evaluating romiplostim to manage thrombocytopenia in MDS was stopped early in 2011 because of data safety monitoring committee concerns regarding the potential for transient increases in blast cell counts and the risk for progression to AML; 15% of romiplostim patients developed greater than 10% blasts compared with 3.6% of placebo patients.83 Preliminary results of continued follow-up of this cohort of patients indicate a similar risk for progression to AML and similar overall survival between romiplostim and placebo patients.84 Romiplostim is also being combined with a DNA hypomethylating agent or lenalidomide in three separate randomized phase II studies to determine its ability to prevent clinically significant thrombocytopenia caused by these agents.85–87 Preliminary data suggest that romiplostim is safe and possibly beneficial when combined with a DNA hypomethylating agent or lenalidomide, but further evaluation is necessary before any conclusions can be drawn about the use of romiplostim in patients with MDS. Eltrombopag is potentially advantageous over romiplostim because it is orally administered. An ongoing phase II trial of eltrombopag administration for management of MDS-related thrombocytopenia will determine its feasibility for use in this patient population. Preliminary results in 17 patients indicate it is safe, is able to increase platelet counts and reduce platelet transfusions, and may improve quality of life.88 The NCCN guidelines do not address use of thrombopoietin-stimulating agents in patients with MDS. Patients should only receive thrombopoietin-stimulating agents under the auspices of a clinical trial until further knowledge is gained about the risk of accelerating the progression to AML.
Transfusion
Patients generally receive RBC transfusions when they develop signs or symptoms of anemia, including tachycardia, fatigue, or dyspnea, which generally occur when hemoglobin drops below 8 to 10 g/dL (4.96 to 6.2 mmol/L).61,89,90 Some clinicians use a transfusion threshold of 10 g/dL (6.2 mmol/L) in patients with significant cardiovascular disease.91 Platelet transfusion is generally reserved for patients with evidence of bleeding to avoid alloimmunization from repeated platelet transfusions, which leads to refractoriness to donor platelets.89,91
Iron Overload
RBC transfusions are associated with shortened leukemia-free and overall survival times in MDS.57,92 It is unclear if this reflects disease severity or is a direct result of iron toxicity.3,93 Recent data indicate MDS patients receiving RBC transfusions more frequently develop infections, cardiac, hepatic, and endocrine dysfunction compared with nontransfused MDS patients or the general population without MDS.12,94 Prospective clinical trials in MDS demonstrate that iron chelation is able to decrease markers of iron overload.95–99 Two studies suggest that patients receiving iron chelation experience hematologic improvement during iron chelation therapy in the absence of other therapy to treat MDS, and three studies suggest iron chelation may improve overall survival.97,98,100–102 In a retrospective study of 18 low- or intermediate-1-risk patients, deferoxamine was associated with improved overall survival compared with matched control participants not receiving iron chelation therapy; the median overall survival time was not reached at 226 months in the deferoxamine group versus 40 months in the control group.100 A prospective cohort of low- or intermediate-1 -risk patients received iron chelation (n = 53) or no iron chelation (n = 44) and was shown to have a median overall survival time of 124 months versus 53 months in nonchelated patients.101 A survival advantage persisted after adjustment for IPSS, age, World Health Organization Classification-based Scoring System (WPSS), level of transfusion requirement, and number of comorbidities. Neukirchen et al. conducted a retrospective, matched-pair analysis of registry data for 94 MDS patients undergoing long-term iron chelation therapy and 94 matched partners in the Düsseldorf MDS Registry; 83% of patients had low- or intermediate-1-risk MDS.102 Patients received supportive care, including growth factors, but no other therapy for MDS other than iron chelation. Median survival time in the iron chelation group was 74 months versus 49 months in the supportive care alone group. It is unclear if the apparent survival benefit with iron chelation is truly a result of reducing iron overload or if confounding factors contributed to these results.103 Preliminary results of a cohort of Medicare beneficiaries with MDS indicate longer duration of deferasirox use correlated with improved overall survival times, but deferasirox was not found to be associated with altered risk of heart failure or endocrine or renal disease.104 It is hypothesized that iron chelation may lower infection risk, improve the outcome of allogeneic HSCT, and delay leukemic transformation in patients with MDS.93 A prospective, randomized trial comparing deferasirox with placebo in low- and intermediate-1-risk MDS patients with transfusional iron overload with a primary outcome of event-free survival is ongoing (registered at www.clinicaltrials.gov; NCT00940602).
The potential toxicity, expense, and benefits of iron chelation should be carefully considered before initiating therapy.103 Deferasirox and deferoxamine are FDA approved for use in patients with chronic iron overload caused by RBC transfusions. Deferiprone is FDA approved for patients with transfusional iron overload secondary to thalassemia when current chelation therapy is inadequate. The prescribing information for deferiprone has a black box warning regarding agranulocytosis, which may lead to serious infection and death. The prescribing information for deferasirox has a black box warning describing renal and hepatic impairment and GI hemorrhage; fatalities were reported. These reactions were more frequently observed in patients with advanced age, high-risk MDS, underlying renal or hepatic impairment, or thrombocytopenia (<50,000 cells/mm3 [50 × 109/L]).



