How, then, does an individual come to have an expanded CAG repeat in his or her HD gene? First, he or she may inherit it from a parent who already has an expanded repeat beyond the normal range but has not yet developed the disease. Second, he or she may inherit an expanded repeat from a parent with repeat length of 35 to 40, which may or may not cause disease in the parent’s lifetime but may expand on transmission, resulting in earlier-onset disease in later generations (and thus explaining anticipation). For example, in the pedigree shown in Figure 7-21, individual I-1, now deceased, was diagnosed with HD at the age of 64 years and was heterozygous for an expanded allele with 37 CAG repeats and a normal, stable allele with 25 repeats. Four of his children inherited the unstable allele, with CAG repeat lengths ranging from 42 to more than 100 repeats. Finally, unaffected individuals may carry alleles with repeat lengths at the upper limit of the normal range (29 to 35 CAG repeats) that can expand during meiosis to 40 or more repeats. CAG repeat alleles at the upper limits of normal that do not cause disease but are capable of expanding into the disease-causing range are known as premutations.
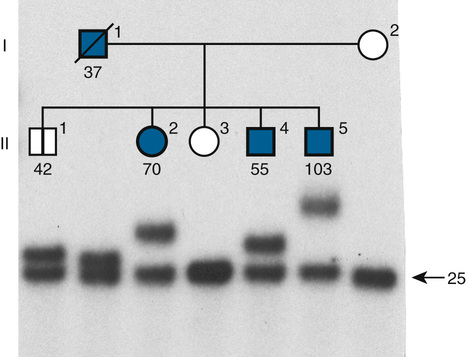
Expansion in HD shows a paternal transmission bias and occurs most frequently during male gametogenesis, which is why the severe early-onset juvenile form of the disease, seen with the largest expansions (70 to 121 repeats), is always paternally inherited.
Fragile X Syndrome
The fragile X syndrome (Case 17) is the most common heritable form of moderate intellectual disability, one of many conditions now considered to be among the autism spectrum disorders. The name fragile X refers to a cytogenetic marker on the X chromosome at Xq27.3, a so-called fragile site induced in cultured cells in which the chromatin fails to condense properly during mitosis. The syndrome is inherited as an X-linked disorder with penetrance in females in the 50% to 60% range. The fragile X syndrome has a frequency of 1 in 3000 to 4000 male births and is so common that it requires consideration in the differential diagnosis of intellectual disability or autism in both males and females. Testing for the fragile X syndrome is among the most frequent indications for genome analysis, genetic counseling, and prenatal diagnosis.
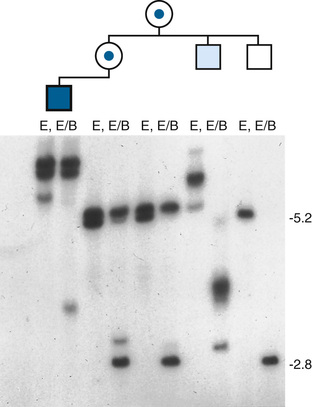
Like HD, fragile X syndrome is caused by an unstable repeat expansion. However, in this case, a massive expansion of a different triplet repeat, CGG, occurs in the 5′ untranslated region of a gene called FMR1 (Fig. 7-22). The normal number of repeats is up to 55, whereas more than 200 (and even up to several thousand) repeats are found in patients with the “full” fragile X syndrome mutation. The syndrome is due to a lack of expression of the FMR1 gene and failure to produce the encoded protein. The expanded repeat leads to excessive methylation of cytosines in the promoter of FMR1; as discussed in Chapter 3, DNA methylation at CpG islands prevents normal promoter function and leads to gene silencing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


