Multiple Sclerosis
KEY CONCEPTS
![]() The etiology of multiple sclerosis (MS) is unknown, but it appears to be autoimmune in nature. Currently there is no cure.
The etiology of multiple sclerosis (MS) is unknown, but it appears to be autoimmune in nature. Currently there is no cure.
![]() MS is characterized by CNS demyelination and axonal damage.
MS is characterized by CNS demyelination and axonal damage.
![]() MS is classified by the nature of progression over time into several categories, which have different clinical presentations and responses to therapy.
MS is classified by the nature of progression over time into several categories, which have different clinical presentations and responses to therapy.
![]() Although studies do not support the general use of any of the FDA-approved disease-modifying therapies (DMTs) in patients with progressive forms of the illness, information derived from multiple studies suggests younger patients with progressive illness and those with either superimposed acute relapses or enhancing lesions on magnetic resonance imaging (MRI) scans may benefit from some of the presently used DMTs.
Although studies do not support the general use of any of the FDA-approved disease-modifying therapies (DMTs) in patients with progressive forms of the illness, information derived from multiple studies suggests younger patients with progressive illness and those with either superimposed acute relapses or enhancing lesions on magnetic resonance imaging (MRI) scans may benefit from some of the presently used DMTs.
![]() Diagnosis of MS requires evidence of dissemination of lesions over time and in multiple parts of the CNS and/or optic nerve, and is made primarily on the basis of clinical symptoms and examination. Diagnostic criteria also allow for the use of MRI, spinal fluid evaluation, optical coherence tomography, and evoked potentials to aid in the diagnosis.
Diagnosis of MS requires evidence of dissemination of lesions over time and in multiple parts of the CNS and/or optic nerve, and is made primarily on the basis of clinical symptoms and examination. Diagnostic criteria also allow for the use of MRI, spinal fluid evaluation, optical coherence tomography, and evoked potentials to aid in the diagnosis.
![]() Exacerbations or relapses of MS can be disabling. When this is the case exacerbations and relapses are treated with high-dose glucocorticoids, such as methylprednisolone IV, with onset of clinical response typically within 3 to 5 days.
Exacerbations or relapses of MS can be disabling. When this is the case exacerbations and relapses are treated with high-dose glucocorticoids, such as methylprednisolone IV, with onset of clinical response typically within 3 to 5 days.
![]() Treatment of relapsing-remitting multiple sclerosis (RRMS) with the DMTs interferon-β (IFN-β) (Avonex, Betaseron, Rebif, Extavia), glatiramer acetate (Copaxone), natalizumab (Tysabri), mitoxantrone (Novantrone), fingolimod (Gilenya), teriflunomide (Aubagio), and dimethyl fumarate (Tecfidera) can reduce annual relapse rate, lessen severity of relapses, slow progression of changes on MRI scans, slow progression of disability, and slow cognitive decline. In addition, they have been shown to reduce the likelihood of developing a second attack after a first clinically isolated syndrome (CIS) consistent with MS.
Treatment of relapsing-remitting multiple sclerosis (RRMS) with the DMTs interferon-β (IFN-β) (Avonex, Betaseron, Rebif, Extavia), glatiramer acetate (Copaxone), natalizumab (Tysabri), mitoxantrone (Novantrone), fingolimod (Gilenya), teriflunomide (Aubagio), and dimethyl fumarate (Tecfidera) can reduce annual relapse rate, lessen severity of relapses, slow progression of changes on MRI scans, slow progression of disability, and slow cognitive decline. In addition, they have been shown to reduce the likelihood of developing a second attack after a first clinically isolated syndrome (CIS) consistent with MS.
![]() In most cases, treatment with DMTs should begin promptly after the diagnosis of relapsing-remitting MS, or after a CIS if the brain MRI is suggestive of high risk of further attacks. Natalizumab and other choices that have been associated with problematic adverse events should be reserved for those patients who have failed one or more standard therapies and those with poor prognostic signs.
In most cases, treatment with DMTs should begin promptly after the diagnosis of relapsing-remitting MS, or after a CIS if the brain MRI is suggestive of high risk of further attacks. Natalizumab and other choices that have been associated with problematic adverse events should be reserved for those patients who have failed one or more standard therapies and those with poor prognostic signs.
![]() The definition of treatment inadequacy for RRMS remains unclear, and therapy changes after “treatment failure” should be individualized.
The definition of treatment inadequacy for RRMS remains unclear, and therapy changes after “treatment failure” should be individualized.
![]() Patients suffering with MS frequently have symptoms such as spasticity, bladder dysfunction, fatigue, neuropathic pain, cognitive dysfunction, and depression that can require treatment. Patients must be counseled that therapies such as IFN-β and glatiramer acetate will not relieve these symptoms. Depression is common in MS and can pose the risk of suicide.
Patients suffering with MS frequently have symptoms such as spasticity, bladder dysfunction, fatigue, neuropathic pain, cognitive dysfunction, and depression that can require treatment. Patients must be counseled that therapies such as IFN-β and glatiramer acetate will not relieve these symptoms. Depression is common in MS and can pose the risk of suicide.
Multiple sclerosis (MS) is an inflammatory disease of the CNS that affects approximately 1 in 200 women and fewer men in the United States.1 The term “multiple sclerosis” refers to two characteristics of the disease: numerous affected areas of the brain and spinal cord (CNS) producing multiple neurologic symptoms that accrue over time, and the characteristic plaques or sclerosed areas that are the hallmark of the disease.
![]() Although MS was first described almost 140 years ago, the cause remains a mystery, and a cure is still unavailable. Nevertheless, many advances have been made in treating and managing the disease complications and improving the quality-of-life of affected individuals.
Although MS was first described almost 140 years ago, the cause remains a mystery, and a cure is still unavailable. Nevertheless, many advances have been made in treating and managing the disease complications and improving the quality-of-life of affected individuals.
EPIDEMIOLOGY
Epidemiologic aspects of MS have been reviewed in many publications.1–5 MS affects approximately 400,000 people in the United States and 2.5 million people worldwide.6 MS is usually diagnosed between the ages of 15 and 45 years; peak incidence occurs in the fourth decade. Approximately 10,000 new cases are diagnosed per year in the United States. Women are afflicted more than men by a ratio of 2:1. Men usually develop the first signs of MS at a later age than women, and are more likely to develop a progressive form of the disease. The most important factors in determination of risk for developing the disease are geography, age, environmental influences, and genetics. In general, disease prevalence is higher the greater the distance from the equator; within the United States the prevalence of MS is higher in states above the 37th parallel. Recent studies, however, suggest a waning latitude gradient as demonstrated by a substantial increase in MS incidence in Mediterranean regions. Rising incidence of MS in females appears to be associated with urbanization. As an example, recent reports suggest that MS incidence markedly rose on Crete among female subjects residing in urban settings or relocating at a young age from rural areas; this suggests that an environmental factor yet to be identified might play a role in changing disease susceptibility.7
MS occurs more frequently in whites of Scandinavian ancestry than in other ethnic groups. In addition, an inverse relationship between MS risk and 25-hydroxyvitamin D levels has been proposed.1,8
Etiology
It is thought that genetically susceptible individuals ≤15 years of age who have lived in a high-risk area for at least 2 years and were exposed to a crucial environmental agent are at risk for developing MS. Interestingly, an individual who migrates from a low- to high-risk area prior to the age of 15 years acquires the same chance of developing MS as those who live in a high-risk area all their lives.2 If the move is made from a high- to a low-risk area, the individual retains the high risk if the move is made after the age of 15 years, but acquires the lower risk if the move is made prior to this age.2 Smoking cigarettes has been associated with both an increased risk of developing MS and with more severe progression of disability.5,9
Viral or bacterial infections may be an important environmental cause of MS. Although no clear association has been identified, certain infections might participate in the pathogenesis of MS by initiating or activating autoreactive immune cells in genetically susceptible individuals, leading to subsequent demyelination. Evidence to support a viral etiology includes increased immunoglobulin G (IgG) synthesis in the CNS, increased antibody titers to certain viruses, and epidemiologic studies that indicate a childhood exposure factor, suggesting that “viral” infections may precipitate exacerbations. In addition, viruses have been shown to cause diseases with prolonged incubation periods, myelin destruction, and a relapsing-remitting course in both humans and experimental animal models.1,10
Although numerous viruses have a proposed association with MS, the greatest evidence supports Epstein–Barr virus (EBV). Links of EBV infection to MS pathology are yet largely hypothetical. Autoreactive T-cells could be activated by EBV through molecular mimicry, whereby sequence similarities between EBV and self-peptides are sufficient to result in the cross-activation of autoreactive T- or B-cells. Other potential mechanisms of demyelination include enhanced breakdown and presentation of self-antigens, expression of viral superantigens, or bystander activation.11 Antibody titers to Epstein–Barr nuclear antigen (EBNA) complex are higher in MS patients versus controls, especially if blood is collected ≥5 years before onset. These titers increase over time in MS patients (controls are unchanged), and a fourfold increase in EBNA titers over time results in a threefold increased risk of developing MS (almost an 18-fold increase in those with first samples before age 20).12 Interestingly, one paper notes individuals positive for HLA DRB1*1501 have a 24-fold increased risk of developing MS when they also have antibodies to certain epitopes within EBNA-1 compared with others.13 This is consistent with a genetic-environmental interaction. In addition, anti-EBNA titers have been associated with relapsing-remitting multiple sclerosis (RRMS), conversion of clinically isolated syndrome (CIS) to clinically definite multiple sclerosis (CDMS, confirmed diagnosis of MS), and with magnetic resonance imaging (MRI) measures such as gadolinium-enhancing lesions, change in T2 lesion volume (r = 0.27; P = 0.044), and Expanded Disability Status Scale (EDSS) score (r = 0.3; P = 0.035). Zivadinov et al. also found anti-EBNA and anti-vascular cell adhesion (VCA) titers associated with gray matter atrophy in MS.14 While Serafini et al. have claimed to identify evidence of abortive infection in a significant number of MS patients,15 others have not been able to replicate these findings.16 The majority of data would lead to a conclusion that exposure to EBV is somehow associated with developing MS, but does not support the concept of an active or aborting EBV infection directly causing MS.
The familial recurrence rate of MS is approximately 5%, with siblings being the most commonly reported relationship,4 and a concordance rate among monozygotic twins of approximately 25%. This is consistent with the idea that an environmental agent is important in the etiology of MS, but also suggests a role for one or more genes. Genes that lie within the major histocompatibility complex (MHC), which is located on the sixth chromosome in humans, have been linked to MS.1,4 Recent data show a significant association of risk with mutations in the interleukin-2α (IL-2α) and interleukin-7α (IL-7α) receptor genes.17–19 African Americans are significantly less likely to be diagnosed with MS compared with whites, although there is emerging evidence that they are more likely to have a severe disease course20 and respond less well to interferon (IFN) therapy.21 A locus on chromosome 1 may be associated with increased susceptibility in African Americans.22
PATHOPHYSIOLOGY
![]() The basic physiologic derangement in MS is stripping of the myelin sheath surrounding CNS axons. This activity is associated with an inflammatory, perivenular infiltrate consisting of T and B lymphocytes, macrophages, antibodies, and complement.10 Demyelination renders axons susceptible to damage, which becomes irreversible when they are severed. Irreversible axonal damage correlates with disability and can be visualized as hypointense lesions, or “black holes,” on T1-weighted MRI.23,24
The basic physiologic derangement in MS is stripping of the myelin sheath surrounding CNS axons. This activity is associated with an inflammatory, perivenular infiltrate consisting of T and B lymphocytes, macrophages, antibodies, and complement.10 Demyelination renders axons susceptible to damage, which becomes irreversible when they are severed. Irreversible axonal damage correlates with disability and can be visualized as hypointense lesions, or “black holes,” on T1-weighted MRI.23,24
It is well accepted that MS lesions are heterogeneous, which may be due in part to differences in the stage of evolution of the lesions over time, differences in underlying immunopathogenesis, or a combination. Briefly stated, acute lesions show demyelination and axonal destruction with lymphocytic activity consistent with an inflammatory state. In contrast, more chronic lesions display less inflammatory lymphocytes with active remyelination.10 Although traditional descriptions have focused on white matter as the sole location of MS lesions, more recent studies have clearly identified cortical and subcortical gray matter lesions both pathologically25 and radiographically.26 In addition, a subset of patients with progressive MS are noted to have abnormalities consistent with B-cell follicles in the meninges.27
Just as the full dimensions of the neuropathology are uncertain, so is the pathogenesis of the MS lesion. Substantial evidence suggests it is an autoimmune process directed against myelin and oligodendrocytes, the cells that make myelin10 (Fig. 39-1). A new concept of T-cell entry into the CNS suggests that the initial lymphocyte invasion in MS may proceed through the ventricles, toward the choroid plexus along a CCL 20 gradient that attracts activated Th17 cells.28 The actual mediator of myelin and axonal destruction has not been established, but may reflect a combination of macrophages, antibodies, destructive cytokines, and reactive oxygen intermediates. The exact trigger for activation of T-cells in the periphery remains unclear, but the T-cells in MS patients recognize myelin basic protein (MBP), proteolipid protein, myelin oligodendrocyte glycoprotein, and myelin-associated glycoprotein. T-helper subtypes can be either pathogenic or protective in MS. Furthermore, theory holds that certain T-cell subsets are not terminally differentiated, but instead engender a level of plasticity that allows for their conversion from pathogenic to protective and vice versa under certain conditions (Fig. 39-2).29 In patients with stable or mild disease, increased numbers of cells are found that express messenger RNA (mRNA) for transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) compared with patients with severe disease. Conversely, a reduction in the number of T-regulatory (Treg) cells, which exhibit suppressor activity, is associated with active MS and can be found in patients with progressive disease. It should be noted, however, that Treg ratios do not always correlate with disease activity. Of note, experimental evidence associates high 25-hydroxyvitamin D levels with improved Treg function, favoring the Th2 phenotype in the Th1/Th2 balance.30 Finally, the significance of one of the immunological hallmarks of MS, the intrathecal synthesis of multiple clones of immunoglobulins, remains unclear. The antigen(s) against which these immunoglobulins are directed remain unknown, but do not appear to include common CNS myelin antigens.31 The complex interplay of a variety of cells, antibodies, and cytokines remains to be elucidated.
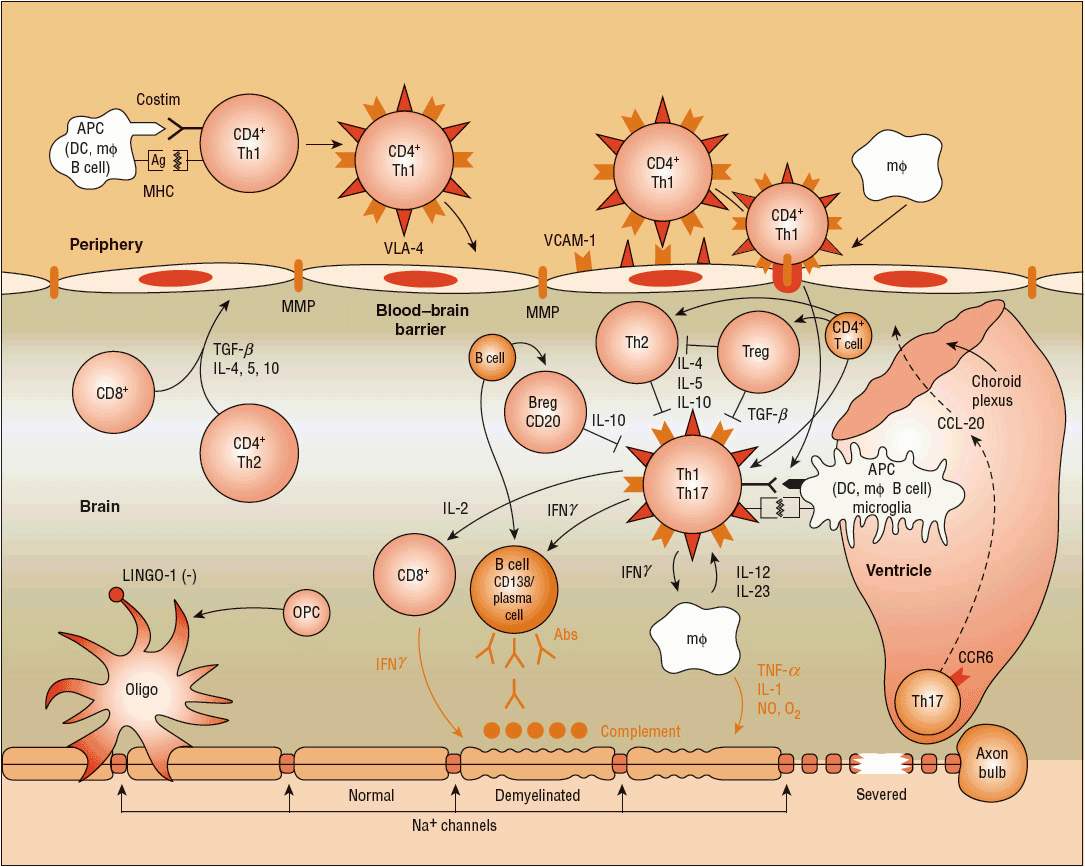
FIGURE 39-1 Autoimmune theory of the pathogenesis of multiple sclerosis (MS). In MS, the immunogenic cells tend to be more myelin-reactive, and these T-cells produce cytokines mimicking a Th1-mediated proinflammatory reaction. T-helper cells (CD4+) appear to be key initiators of myelin destruction in MS. These autoreactive CD4+ cells, especially of the T-helper cell type 1 (Th1) subtype, are activated in the periphery, perhaps following a viral infection. The activation of T- and B-cells requires two signals. The first signal is the interaction between MHC and APC (macrophage, dendritic cell, B-cell). The second signal consists of the binding between B7 on the APC and CD28 on the T-cell for T-cell activation. Similarly, CD40 expressed on APCs and CD40L expressed on T-cells interact to signal the proliferation of B-cells within the blood–brain barrier following the entry to T-cells. The T-cells in the periphery express adhesion molecules on their surfaces that allow them to attach and roll along the endothelial cells that constitute the blood–brain barrier. The activated T-cells also produce MMP that help to create openings in the blood–brain barrier, allowing entry of the activated T-cells past the blood–brain barrier and into the CNS. Once inside the CNS, the T-cells produce proinflammatory cytokines, especially interleukins (ILs) 1, 2, 12, 17, and 23, tumor necrosis factor-α (TNF-α), and interferon-γ (INF-γ), which further create openings in the blood–brain barrier, allowing entry of B-cells, complement, macrophages, and antibodies. The T-cells also interact within the CNS with the resident microglia, astrocytes, and macrophages, further enhancing production of proinflammatory cytokines and other potential mediators of CNS damage, including reactive oxygen intermediates and nitric oxide. The role of modulating, or downregulating, cytokines such as IL-4, IL-5, IL-10, and transforming growth factor-β (TGF-β) also has been described. These cytokines are the products of CD4+, CD8+, and Th1-cells.10 New pathogenic mechanisms involve, but are not limited to, receptor-ligand mediated T-cell entry via choroid plexus (CCR6-CCL20 axis),28 coupling of key receptor-ligands for inhibition of myelination/demyelination (LINGO-1/NOGO66/p75 or TROY complex, Jagged-Notch signaling).(Ag, antigens; APC, antigen presenting cell; DC, dendrite cell; IgG, immunoglobulin G; MΦ, macrophage; Na+, sodium ion; MMP, matrix metalloproteinases; MHC, major histocompatibility complex; OPC, oligodendrocyte precursor cell; VLA, very late antigen; VCAM, vascular cell adhesion molecule.)
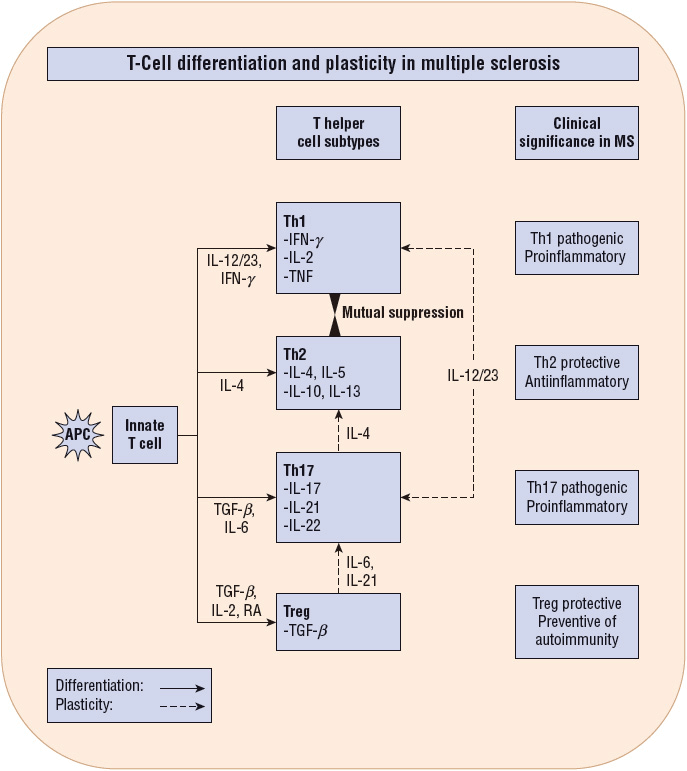
FIGURE 39-2 Upon interaction with an antigen-laden APC and specific cytokines, the innate T-cells undergo differentiation into a few lineages (subtypes). Four subtypes significant for MS pathophysiology are illustrated here (Th1, Th2, Th17, and Treg). Th1 and Th17 are proinflammatory, Th2 is anti-inflammatory, and Treg is regulatory. Th1 and Th2 are mutually suppressive and are relatively stable differentiated subtypes. In contrast, Th17 and Treg subtypes are recently found to exhibit “plasticity.” In other words, they can undergo phenotypic conversion to another T-cell subtype (Th1 or Th2) in the presence of specific cytokine conditions. This plasticity of Th17 and Treg is the immunologic basis for development of therapeutic agents to favor the production of suitable Th subtypes for combating microbial invasion and also concurrently achieving neurocellular recovery after an infection.29 (APC, antigen presenting cell.)
CLINICAL PRESENTATION Multiple Sclerosis
CLINICAL PRESENTATION AND COURSE OF ILLNESS
![]() The clinical presentation of MS is extremely variable among patients and typically varies over time in a given patient. The signs and symptoms of MS can be divided into three categories. Primary symptoms are a direct consequence of conduction disturbances produced by demyelination and axonal damage, and reflect the area of the CNS that is damaged. Secondary symptoms are complications resulting from primary symptoms. For example, urinary retention, a primary symptom, can lead to frequent urinary tract infections (UTIs), a secondary symptom. Tertiary symptoms relate to the effect of the disease on the patient’s everyday life.32
The clinical presentation of MS is extremely variable among patients and typically varies over time in a given patient. The signs and symptoms of MS can be divided into three categories. Primary symptoms are a direct consequence of conduction disturbances produced by demyelination and axonal damage, and reflect the area of the CNS that is damaged. Secondary symptoms are complications resulting from primary symptoms. For example, urinary retention, a primary symptom, can lead to frequent urinary tract infections (UTIs), a secondary symptom. Tertiary symptoms relate to the effect of the disease on the patient’s everyday life.32
The clinical course of CDMS is classified into four categories.33 At the onset of symptoms, about 85% of patients have exacerbations—new symptoms lasting at least 24 hours and separated from other new symptoms by at least 30 days—followed by remissions (complete or incomplete). Exacerbations are frequently referred to as relapses or attacks. This course is called RRMS; the first clinical presentation is typically CIS. During the RRMS phase, there is a correlation between new brain MRI lesions and clinical attacks, but typically there are many more new MRI lesions than new clinical symptoms. In RRMS patients, attack frequency tends to decrease over time and becomes independent of the development of progressive disabilities.34 Neurologic recovery following an exacerbation is often quite good early in the disease course, but following repeated relapses, recovery tends to be less complete. In addition, there is a new concept of a radiologically isolated syndrome (RIS), referring to individuals who have clinical scenarios not typical of MS, yet obtain MRI scans for other reasons (e.g., headache) and have radiological signs suggestive of MS. Some percentage of these patients convert to RRMS over time,35 although when to start treatment remains unclear and varies by practice.
Up to 10% to 20% of RRMS patients have a benign course, characterized by few relapses, often sensory, with minimal disability accruing over time. Most RRMS patients eventually enter a progressive phase in which attacks and remissions are difficult to identify. This is referred to as secondary-progressive multiple sclerosis (SPMS). Disability tends to accumulate more significantly during this phase of the illness. New brain MRI lesions, especially those seen only after the injection of contrast material, are less common, and brain atrophy and T1 holes increase.36
![]() Approximately 15% of patients never have attacks and remissions but have progressive disease from the outset, known as primary-progressive multiple sclerosis (PPMS). These patients will have symptoms, especially spastic paraparesis that may worsen rapidly or relatively slowly over time, and accrue progressively more disability. Patients with PPMS are diagnosed at a later age, with the number of males roughly equal to that of females. In general, PPMS patients tend to have a worse prognosis than those who present initially with RRMS, although more recent data suggest progression is variable.37 Many clinical trials have suggested that a significant portion of patients with PPMS do not receive benefit from studied therapies. However, a recent article using rituximab suggests a subgroup of PPMS patients who are <51 years of age and have at least one gadolinium-enhancing lesion may benefit from this therapy.38 Finally, a small percentage of patients may have a mixture of both progression and relapses, referred to as progressive-relapsing multiple sclerosis (PRMS). These patients are generally treated as relapsing patients.
Approximately 15% of patients never have attacks and remissions but have progressive disease from the outset, known as primary-progressive multiple sclerosis (PPMS). These patients will have symptoms, especially spastic paraparesis that may worsen rapidly or relatively slowly over time, and accrue progressively more disability. Patients with PPMS are diagnosed at a later age, with the number of males roughly equal to that of females. In general, PPMS patients tend to have a worse prognosis than those who present initially with RRMS, although more recent data suggest progression is variable.37 Many clinical trials have suggested that a significant portion of patients with PPMS do not receive benefit from studied therapies. However, a recent article using rituximab suggests a subgroup of PPMS patients who are <51 years of age and have at least one gadolinium-enhancing lesion may benefit from this therapy.38 Finally, a small percentage of patients may have a mixture of both progression and relapses, referred to as progressive-relapsing multiple sclerosis (PRMS). These patients are generally treated as relapsing patients.
Progression of the illness throughout the lifetime can be measured in many ways. The most widely used clinical rating scale is the EDSS, which uses a numerical value ranging from 0 (no disability) to 10 (death) to evaluate neurologic functions.39 The limitations of this scale are the relative insensitivity to clinical changes not involving impairment of ambulation, such as changes in cognition, fatigue, and affect. Other tools, such as the multiple sclerosis functional composite (MSFC), are being evaluated for increased sensitivity and utility in describing changes in MS-related disability over time.40 Increasingly, MRI is being used as an index of both disease activity and progression.10 Specifically, the appearance of new lesions or changes in lesion number, size, and volume are being used as outcome measures in research studies. Optical coherence tomography measures the retinal neural fiber layer thickness, and may also be a measurable sign of pathological progression over time.41
The unpredictable nature of MS makes it impossible to anticipate when an exacerbation will occur. However, certain factors, including infections, heat (including fever), sleep deprivation, stress, malnutrition, anemia, concurrent organ dysfunction, exertion, and childbirth, may aggravate symptoms or lead to an attack. Interestingly, many patients experience a significant reduction in relapses during the third trimester of pregnancy, followed by a relative increase postpartum.42 Between 60% and 80% of individuals diagnosed with the MS have been reported to be sensitive to environmental heat.
Clinically, increased body temperature might result in worsening of previous neurological deficits, including fatigue and decreased muscular endurance. Blurred vision, known as Uthoff’s phenomenon, is caused by increased body temperature due to physical exercise or physical restraint. Body temperature influences nerve impulses, which are blocked or slowed down in a damaged nerve. After normalization of the temperature, signs and symptoms improve or disappear.
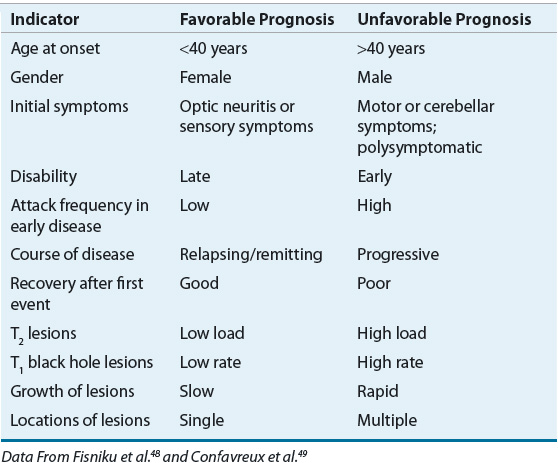
MS usually does not directly diminish life expectancy. The development of secondary complications such as pneumonia or septicemia (secondary to aspiration in those with swallowing difficulties, decubitus ulcers, or UTIs) or rapid progression of primary lesions affecting respiratory function can lead to a shorter than expected life span. Most of the decrease in life span is seen in patients with rapidly progressive disease. Suicide rates as high as seven times that seen in the general population have been reported.43 Clinical and demographic factors used to predict prognosis of MS are listed in Table 39-1.5,44 Several MRI features also have been shown to correlate with progression of disease (see below).45–47
TABLE 39-1 Prognostic Indicators in Multiple Sclerosis
DIAGNOSIS
![]() MS is a diagnosis of exclusion; symptoms frequently can be attributed to other neurologic diseases, just as many syndromes can mimic MS. Some patients may have typical symptoms consistent with classic CIS, whereas others may have symptoms that are more vague. The diagnosis remains primarily a clinical one that requires demonstration of “lesions separated in space and time,” referring to the occurrence of at least two episodes of neurologic disturbance reflecting distinct sites of CNS damage that cannot be explained by another mechanism.50 An international panel of MS experts established the McDonald criteria,50 which allows brain MRI lesions, cerebrospinal fluid (CSF) abnormalities, and visual-evoked potential (VEP) studies to substitute for clinical lesions in defining “separated in space and time.” A reevaluation of the McDonald criteria has simplified the use of these laboratory studies.45 In the new scheme, diagnostic categories are MS, possible MS (for those individuals at high risk of developing MS), and not MS; these new criteria allow for earlier diagnosis.45 Newer, simpler MRI criteria defining dissemination in time and space may be somewhat more sensitive and equally specific.51–53 A consensus panel of the American Association of Neurology endorses the utility of MRI for diagnostic purpose,47 and the U.S. FDA has approved several of the immunotherapies to be used after a single attack (CIS) of demyelination in the context of an appropriately abnormal brain MRI. A proposed set of criteria now being considered will allow for earlier diagnosis in patients with CIS to establish “dissemination in time and space” with a single MRI. Therefore, patients will need to have lesions in different areas of their CNS with at least one enhancing lesion that correlates with clinical symptomatology. By fulfilling these criteria, a patient can be diagnosed with CDMS.
MS is a diagnosis of exclusion; symptoms frequently can be attributed to other neurologic diseases, just as many syndromes can mimic MS. Some patients may have typical symptoms consistent with classic CIS, whereas others may have symptoms that are more vague. The diagnosis remains primarily a clinical one that requires demonstration of “lesions separated in space and time,” referring to the occurrence of at least two episodes of neurologic disturbance reflecting distinct sites of CNS damage that cannot be explained by another mechanism.50 An international panel of MS experts established the McDonald criteria,50 which allows brain MRI lesions, cerebrospinal fluid (CSF) abnormalities, and visual-evoked potential (VEP) studies to substitute for clinical lesions in defining “separated in space and time.” A reevaluation of the McDonald criteria has simplified the use of these laboratory studies.45 In the new scheme, diagnostic categories are MS, possible MS (for those individuals at high risk of developing MS), and not MS; these new criteria allow for earlier diagnosis.45 Newer, simpler MRI criteria defining dissemination in time and space may be somewhat more sensitive and equally specific.51–53 A consensus panel of the American Association of Neurology endorses the utility of MRI for diagnostic purpose,47 and the U.S. FDA has approved several of the immunotherapies to be used after a single attack (CIS) of demyelination in the context of an appropriately abnormal brain MRI. A proposed set of criteria now being considered will allow for earlier diagnosis in patients with CIS to establish “dissemination in time and space” with a single MRI. Therefore, patients will need to have lesions in different areas of their CNS with at least one enhancing lesion that correlates with clinical symptomatology. By fulfilling these criteria, a patient can be diagnosed with CDMS.
Laboratory Studies
To date, there are no tests specific for MS. Evidence provided by MRI of the brain and spine,46,47 CSF evaluation (presence of increased oligoclonal bands and increased IgG), evoked potentials,45,50 and optic coherence tomography,54 used in conjunction with the physical examination and history, aids in establishing the diagnosis of MS. MRI, the most valuable diagnostic tool, produces images of the brain and spine that reflect damage that is characteristic of MS plaques in multiple areas of the CNS. MRI is the preferred technique for establishing a diagnosis, prognosis, and for following disease progression. Optic neuritis, a lesion or lesions on the optic nerve, is a common first symptom of MS. A greater number of T2-weighted lesions (called T2 burden of disease) on MRI following optic neuritis or CIS appears to correlate with the development of disability and progression to CDMS.46 Lesions that enhance after injection of the contrast material gadolinium indicate new lesions and disruption of the blood–brain barrier and are associated with early conversion to CDMS in CIS patients.46,55 However, they do not correlate well over time with progression of disability. Brain atrophy, even early in the course of the illness, probably correlates better with progression of disability.47
DIFFERENTIAL DIAGNOSIS
Because a number of disorders can mimic MS, most patients are screened with blood tests for rheumatologic, collagen-vascular, infectious, and sometimes inherited metabolic diseases. Electromyography may help in diagnosing amyotrophic lateral sclerosis and neuropathies.
MRI, used to rule out tumors and cervical spondylosis, may also lead to evaluations for MS in many patients with little or no clinical history of MS. While some of these patients may have MRI scans suggestive of MS (so-called RIS), most have nonspecific MRI scans with identifiable causes for their scan abnormalities, including age greater than 50 years, hypertension, and migraine.56 The use of established criteria for distinguishing MS lesions from other etiologies enhances diagnostic accuracy.
TREATMENT
Treatment of MS falls into three broad categories: treatment of exacerbations, disease-modifying therapies (DMTs), and symptomatic therapies. Treatment of exacerbations will shorten the duration and possibly decrease the severity of the attack. DMTs alter the course of the illness, and diminish progressive disability over time. Symptomatic management of the disease is of utmost importance to maintain the patient’s quality-of-life. Although different treatment modalities have been studied in the last 30 years, many older trials had flawed designs. As there are no universally accepted treatment algorithms, treatments vary among clinicians and centers. Perhaps more importantly, treatment decisions are frequently based on the wishes and goals of individual patients rather than evidence-based algorithms. One potential algorithm for the immunotherapy of CDMS is shown in Figure 39-3.
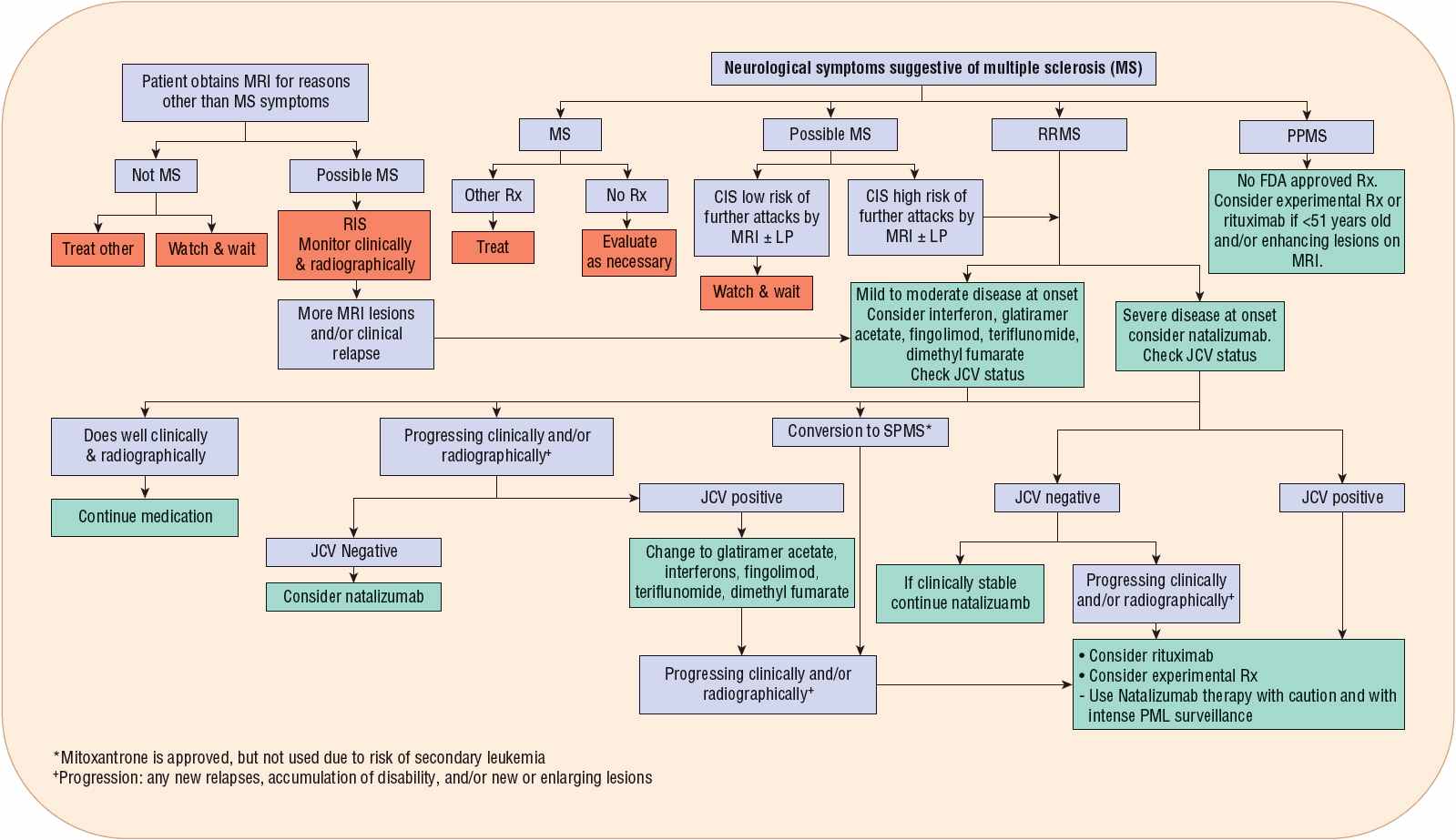
FIGURE 39-3 Algorithm for management of clinically definite multiple sclerosis. (ABC-R, interferon β1a [Avonex], interferon β1b [Betaseron, Extavia], glatiramer acetate [Copaxone], and interferon β1a [Rebif]; IVIG, intravenous immunoglobulin.)
Desired Outcomes
The main goals of treatment are to improve patients overall quality-of-life and minimize long-term disability. Treatment goals are attained by altering MS exacerbations or relapses, decreasing the number of white matter lesions and black holes on MRI, averting brain atrophy, and ultimately halting disease progression. This can be achieved by early recognition of the disease (CIS) and immediate utilization of FDA-approved drugs.
General Approach to Treatment
The severity of symptoms at initial presentation will determine whether an induction or escalation algorithm will be assigned to an individual patient. When FDA-approved drugs do not alter the naturally progressive disease, investigational agents or non–FDA-approved medications, such as rituximab, may be used. As a general rule, MS affects patients in their most productive years of life. Practitioners must work with their patients to set realistic expectations over their lifetime and develop a long-term treatment and management plan. Patients may require external support and assistance to accept their diagnosis. With disease progression, patients are likely to acquire secondary and tertiary symptoms of MS. In clinical trials, high nonadherence rates are reported as an important issue for potential treatment failure. Potential reasons identified for nonadherence are lack of perceived benefit, cost, adverse effects, depression, and fear of needles. With proper patient education and therapy management, treatment failure due to nonadherence can be avoided. Specialty pharmacies may be useful to address patient concerns. With the advance of FDA-approved medications to treat MS, patients are experiencing fewer relapses, slower disease progression, and improved quality-of-life.
Treatment of Exacerbations
![]() Exacerbations are the hallmark of early RRMS. Although recovery after relapses is in general complete, over time a substantial accumulation of disability occurs. Controversy exists about the relationship between relapses and subsequent accumulation of disability. Frequent relapses (more than three relapses per year in the first 2 years after diagnosis), particularly in early phases of the disease, have shown consistent positive correlation with later development of neurological disability. Generally, mild exacerbations that do not produce functional decline may not require treatment. Decisions to treat relapses are usually substantiated by patient’s expectations, prior experience with corticosteroids, and predicted course of recovery. Generally accepted indications are based on mono- or polysymptomatic presentations; relapses that localize to the optic nerve, spinal cord, or brainstem; functional limitations that affect activities of daily living; and symptoms that continue to worsen over a period of 2 weeks. When functional ability is affected, the standard intervention is IV injection of high-dose corticosteroids. The American Academy of Neurology recommends that if treatment with steroids is warranted, it is best to use IV methylprednisolone.57 The mechanism of action for corticosteroids in MS is unknown, but it is speculated that steroids improve recovery by decreasing edema in the area of demyelination. IV methylprednisolone has been shown to shorten the duration of exacerbations; it may also delay repeat attacks for up to 2 years after optic neuritis,57 although it has not been shown to definitively affect disease progression.58 More recently, some practitioners are using high doses of oral methylprednisolone, mixing the lyophilized powder or crushed oral tablets in flavored drinks such as smoothies, but there are no comparative data to establish that this is an equivalent way to deliver the medication. In some circumstances, equipotent doses of oral prednisone can be substituted for IV methylprednisolone. Interestingly, adrenocorticotropic hormone (ACTH) is the only agent that is FDA approved for treatment of MS exacerbation treatment, although it is rarely used due to cost and availability.
Exacerbations are the hallmark of early RRMS. Although recovery after relapses is in general complete, over time a substantial accumulation of disability occurs. Controversy exists about the relationship between relapses and subsequent accumulation of disability. Frequent relapses (more than three relapses per year in the first 2 years after diagnosis), particularly in early phases of the disease, have shown consistent positive correlation with later development of neurological disability. Generally, mild exacerbations that do not produce functional decline may not require treatment. Decisions to treat relapses are usually substantiated by patient’s expectations, prior experience with corticosteroids, and predicted course of recovery. Generally accepted indications are based on mono- or polysymptomatic presentations; relapses that localize to the optic nerve, spinal cord, or brainstem; functional limitations that affect activities of daily living; and symptoms that continue to worsen over a period of 2 weeks. When functional ability is affected, the standard intervention is IV injection of high-dose corticosteroids. The American Academy of Neurology recommends that if treatment with steroids is warranted, it is best to use IV methylprednisolone.57 The mechanism of action for corticosteroids in MS is unknown, but it is speculated that steroids improve recovery by decreasing edema in the area of demyelination. IV methylprednisolone has been shown to shorten the duration of exacerbations; it may also delay repeat attacks for up to 2 years after optic neuritis,57 although it has not been shown to definitively affect disease progression.58 More recently, some practitioners are using high doses of oral methylprednisolone, mixing the lyophilized powder or crushed oral tablets in flavored drinks such as smoothies, but there are no comparative data to establish that this is an equivalent way to deliver the medication. In some circumstances, equipotent doses of oral prednisone can be substituted for IV methylprednisolone. Interestingly, adrenocorticotropic hormone (ACTH) is the only agent that is FDA approved for treatment of MS exacerbation treatment, although it is rarely used due to cost and availability.
Methylprednisolone doses range from 500 to 1,000 mg/day, given IV. Duration of therapy is variable and can range from 3 to (rarely) 10 days, depending on clinical response. Functional recovery after an exacerbation is more rapid if corticosteroids are initiated within 2 weeks of symptom onset. If improvement occurs, it usually begins after 3 to 5 days. Short-term use is often accompanied by sleep disturbance, a metallic taste, and rarely, GI upset. Patients with diabetes mellitus or a predilection to diabetes mellitus may have significant elevations of blood sugar, requiring the use of insulin. Longer durations of IV methylprednisolone therapy are associated with acne and fungal infections, mood alteration, and rarely, GI hemorrhage (especially in hospitalized patients or in those taking aspirin). If methylprednisolone is not available, equipotent doses of dexamethasone have been used as a substitute, although this is not well supported in the literature.
A small number of patients have more severe attacks, manifested by hemiplegia, paraplegia, or quadriplegia. If these patients fail to improve with aggressive steroid therapy, plasma exchange every other day for seven treatments can be beneficial for approximately 40% of patients, or intravenous immunoglobulin (IVIG) can be given.
A “pseudo-exacerbation” is an episode with symptoms consistent with an exacerbation, but precipitated by something other than the natural course of the disease. A pseudo-exacerbation can be precipitated by heat, infections (e.g., UTIs), or stress (emotional or physical); these must be ruled out before treatment is initiated or DMTs altered.
Disease-Modifying Therapy
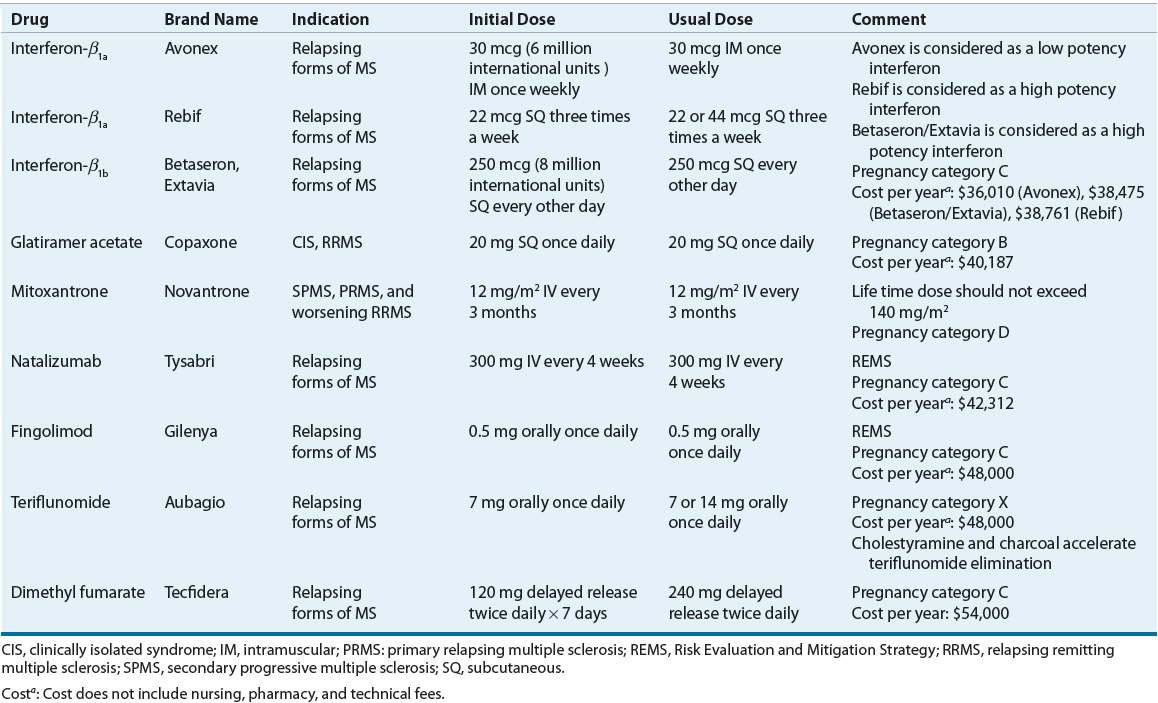
![]() Indications and dosing of DMTs is shown in Table 39-2. MS is a complex, heterogeneous disease with clear variability in pathogenesis between patients and within patients over time. As a result, treatment decisions are usually based on clinical predictors of disease severity, our incomplete understanding of the mechanism of action of currently available therapies, and the safety and tolerability profile of the medications. There is some degree of agreement that use of escalation approaches early in the course of the disease, with safer yet partially effective medications, is useful. These concepts lead to various categories of therapies: first-, second-, and potentially third-line medications. Currently, FDA-approved first-line therapies (self-injected medications that decrease annualized relapse rate by about 30% and decrease the formation of new white matter lesion) include three IFN formulations (four brand names), and glatiramer acetate (a non-IFN). The first-line DMTs are not immediately efficacious for patient symptoms. However, their efficacy is noted approximately 1 to 2 years after starting therapy. Fingolimod, natalizumab, and mitoxantrone, also approved for the treatment of MS patients, are used in cases of inadequate response or intolerance to first-line agents. The FDA has approved natalizumab, fingolimod, teriflunomide, and dimethyl fumarate for the treatment of relapsing forms of MS. Mitoxantrone has an FDA indication for progressive or worsening MS.
Indications and dosing of DMTs is shown in Table 39-2. MS is a complex, heterogeneous disease with clear variability in pathogenesis between patients and within patients over time. As a result, treatment decisions are usually based on clinical predictors of disease severity, our incomplete understanding of the mechanism of action of currently available therapies, and the safety and tolerability profile of the medications. There is some degree of agreement that use of escalation approaches early in the course of the disease, with safer yet partially effective medications, is useful. These concepts lead to various categories of therapies: first-, second-, and potentially third-line medications. Currently, FDA-approved first-line therapies (self-injected medications that decrease annualized relapse rate by about 30% and decrease the formation of new white matter lesion) include three IFN formulations (four brand names), and glatiramer acetate (a non-IFN). The first-line DMTs are not immediately efficacious for patient symptoms. However, their efficacy is noted approximately 1 to 2 years after starting therapy. Fingolimod, natalizumab, and mitoxantrone, also approved for the treatment of MS patients, are used in cases of inadequate response or intolerance to first-line agents. The FDA has approved natalizumab, fingolimod, teriflunomide, and dimethyl fumarate for the treatment of relapsing forms of MS. Mitoxantrone has an FDA indication for progressive or worsening MS.
TABLE 39-2 Disease Modifying Therapy