Molecular Diagnosis
New insights during the last two decades have catapulted genetic studies of malignant lymphomas from a research tool to a routine laboratory test. Unlike immunophenotypic studies using flow cytometry or immunohistochemistry, which depend on protein expression, molecular testing directly analyzes the DNA of genes encoding immunoglobulins (Igs) and T-cell receptors (TCRs) that rearrange during B- and T-cell development. Molecular methods can also directly analyze the DNA of oncogenes and tumor suppressor genes involved in lymphomagenesis (1,2,3,4,5).
To provide a framework for readers with different backgrounds, this chapter emphasizes the strategies for molecular diagnostic tests commonly used for the workup of malignant lymphomas, and is particularly focused on antigen receptor gene rearrangements and chromosomal translocations. We also compare the relative advantages and disadvantages of using Southern blot hybridization (also known as restriction fragment-length polymorphism), polymerase chain reaction (PCR), conventional cytogenetics, and fluorescence in situ hybridization (FISH) analysis.
Antigen Receptor Genes
The human immune system can recognize an almost unlimited variety of foreign antigens. This is achieved by the creation of millions of Ig and TCR molecules. Because Igs and TCRs are proteins, and genes encode proteins, the information for these molecules must be stored in the DNA. The body has evolved efficient mechanisms to generate this diversity. The Ig and TCR genes, which belong to the same antigen receptor supergene family, are structurally and functionally similar. These genes are composed of discontinuous segments that encode for the variable (V), joining (J), and constant (C) regions of Igs and TCRs (Fig. 8.1). Some of genes also contain diversity (D) region gene segments. By shuffling and recombining (analogous to shuffling a deck of playing cards) the few hundred discontinuous gene segments belonging to the Ig and TCR gene families, each human being can theoretically produce millions of complete antigen receptor molecules (6).
This process of Ig and TCR gene rearrangement utilizes similar consensus DNA sequences adjacent to V, D, and J segments and the same recombinase enzymes that facilitate rearrangement (7,8). For a lymphoid progenitor cell to become a B or T cell, it must rearrange its Ig or TCR genes, respectively. The process of VDJ or VJ rearrangement occurs in the bone marrow. This shuffling of gene segments accounts, in large part, for the diversity of the immune system at the protein level. However, other mechanisms further explain the diversity of the immune system.
Generation of Diversity
At least six molecular mechanisms, operating at different stages of lymphoid differentiation, act synergistically to generate the enormous antigen-recognizing capacity of the immune system: (a) the presence of noncoding DNA sequences, called introns, which separate the protein-coding DNA sequences, called exons, of the V, D, J, and C antigen receptor gene segments; (b) somatic recombination; (c) sharing of junctional nucleotides donated from contiguously rearranged gene segments (e.g., part V and part J) to create a new hybrid codon, which can alter the amino acid sequence; (d) somatic mutation; (e) derivation of multiple proteins from a single gene, either with multiple transcriptional promoters or with different exons that can be alternatively spliced into or out of the messenger RNA; and (f) combinatorial association of polypeptide subunits.
Somatic Recombination
Somatic recombination of the antigen receptor genes is a step-by-step process in which the deleting and joining of discontinuous gene segments creates a new linear genetic code that differs substantially from the embryonic or germline DNA (Fig. 8.2). Heptamer–nonamer sequences flanking the gene segments are used in the process of recombination, and the recombinase enzymes, RAG1 and RAG2, are required. Because the probability of two cells rearranging their Ig or TCR genes identically is low, the DNA of each lymphocyte is therefore unique. Imprecise gene segment alignment further diversifies the immune repertoire. Deleting nucleotides via exonucleases and inserting non–template-dependent nucleotides (N insertion) via terminal deoxynucleotidyl transferase (TdT), especially at the Ig heavy chain (IgH) D junctions, generates further diversity.
Complementarity-Determining Regions
Junctional alterations help refine the specificity of the antigen-binding domain, also called the complementarity-determining regions (CDRs), by producing hypervariable DNA sequences at VDJ and VJ joints. At a later stage of maturation, antigen-stimulated, germinal-center memory B cells undergo somatic hypermutation of their CDRs to produce high-affinity antibodies. In contrast, TCRs rarely undergo somatic mutation. These genetic mechanisms irreversibly alter the DNA to produce a molecular signature for each B and T cell.
In-Frame Alignment of Gene Segments
Exact, in-frame alignment of several widely separated exons is required for successful Ig gene rearrangement. This process is prone to failure. In particular, faulty gene segment joints can disrupt the DNA coding sequence and lead to improperly located stop codons or missense codons. Only about one-third of V to J light chain gene segments rearrange correctly in-frame. This agrees with predictions based on genetic code syntax, because a nucleotide triplet determines each amino acid. Even fewer Ig heavy chains align in-frame, because they depend on two consecutive events: D to J followed by V to DJ rearrangement. An interesting difference between Ig and TCR genes is that IgH D gene segments have only one open reading frame, whereas TCRδ D gene segments are readable in all three frames.
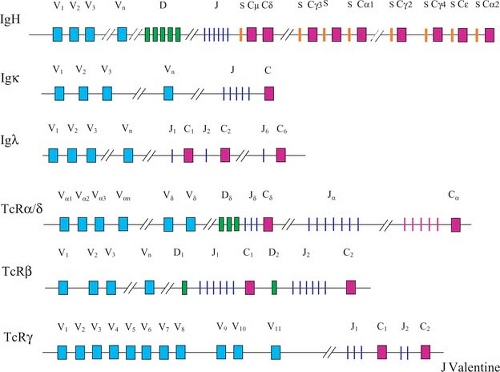 Figure 8.1. Schematic diagram showing the germline configuration of the antigen receptor genes. |
Genetic Hierarchy
During early, antigen-independent lymphoid differentiation, normal B- and T-cell precursors rearrange their Ig and TCR genes in hierarchical fashion. The usual early to late rearrangement order for B cells is IgH, followed by the Igκ and then Igλ light chain genes. For T cells, the TCRδ chain gene rearranges first, followed by the TCRγ, TCRβ, and then TCRα genes (9,10).
Allelic Exclusion
Immunoglobulin molecules have a basic heterodimeric structure: Ig heavy chain plus Ig light chain. Depending on the Ig heavy chain class, these heterodimers then form dimers (IgG) or oligomers (IgM) to produce a fully functional Ig antigen receptor. If one IgH allele rearranges its gene segments in such a fashion as to preclude encoding a complete Ig heavy chain (referred to as a nonproductive gene rearrangement), then the other IgH allele rearranges. A similar rule applies to the Ig light chain loci, with the additional requirement that Igκ gene rearrangement precedes Igλ gene rearrangement. Therefore, in a normal B-cell precursor, the Igκ gene rearranges first; only if both alleles rearrange nonfunctionally can the Igλ gene then rearrange. This is known as the principle of allelic exclusion (11).
All productive and nonproductive gene rearrangement attempts are permanently recorded in the DNA and can be detected by Southern blot analysis. In the most extreme case (e.g. an IgM λ-bearing B cell), both IgH alleles, both κ alleles, and both λ alleles could rearrange before yielding functional rearrangements that allow transcription and translocation of a functional Ig molecule.
As immature T cells transit from the cortex to medulla of the thymus, positive and negative selection determines T-cell survival and exportation to the peripheral lymphoid organs. The expression of the TCR plays a central role; clones detecting a single antigen–major histocompatability complex interaction live, and clones recognizing self-antigens die. Individual mature T cells express either TCRαβ (95% of T cells) or TCRγδ (5% of T cells), but not both (10).
Multiple Constant Regions and Class Switching
The VDJ (or VJ) DNA regions encoding the antigen-binding portion of the IgH may recombine with more than one C region. This process is known as class switching, and represents another gene rearrangement process. In B cells, exposure to antigen in the germinal centers of secondary follicles leads to class switching by deleting Cμ (and often Cδ) and rearranging VDJ next to one of the other Ig heavy chains (e.g., Cγ, Cα, or Cε). These deletions can occur via switch regions adjacent to the C regions (except Cδ).
Molecular Diagnosis of Clonality
Molecular testing is the gold standard for diagnosing B- or T-cell clonality (1,2,3,4). The clinical laboratory routinely determines lymphocyte clonality either by Southern blot hybridization or by PCR. Although molecular diagnostic testing for clonal antigen receptor gene rearrangements is highly specific, a number of biologic and technical factors limit the interpretation of results. Importantly, only lymphoid cells that rearrange their genes can be assessed for gene rearrangements. Therefore, true natural killer (NK) cells maintain their antigen receptor gene segments in a germline configuration, and NK-cell
derived lymphomas do not carry antigen receptor gene rearrangements. Another important caveat is that a variety of reactive lymphoid lesions may contain detectable clones (12,13). Because clonality is not synonymous with malignancy, molecular tests should be correlated with clinical, morphologic, and immunophenotypic data.
derived lymphomas do not carry antigen receptor gene rearrangements. Another important caveat is that a variety of reactive lymphoid lesions may contain detectable clones (12,13). Because clonality is not synonymous with malignancy, molecular tests should be correlated with clinical, morphologic, and immunophenotypic data.
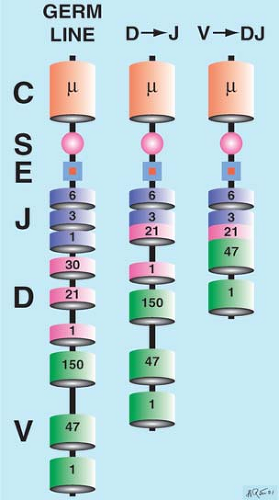 Figure 8.2. Somatic recombination. Immunoglobulin heavy chain gene segments rearrange stepwise from germline (left), to D-J (center), to VDJ (right). The combinatorial possibilities of 150 Vs, 30 Ds, and six Js are suggested by this example. D-21 moves adjacent to J-3, deleting DNA sandwiched between (center). Then, V-47 links to D-21, removing any connecting DNA (right) and completing a unique VDJ recombination. C, constant; S, switch region; E, enhancer; J, joining; D, diversity; V, variable. (Figure courtesy of H. Ratech, M.D.) |
Southern blot analysis to detect antigen receptor gene rearrangements requires high-molecular-weight genomic DNA as starting material. In most laboratories, this means freezing and storing fresh lymph node tissue at extremely cold temperatures. Buffers for lysis, storage, and transportation also have been developed that promise to facilitate tissue processing by preserving lymph node samples up to 4 weeks at room temperature for genomic DNA Southern blotting or PCR analysis (14).
Southern Blot Analysis of Antigen Receptor Genes
Immunoglobulin Heavy Chain Gene
The IgH gene at chromosome 14q32 includes clusters of multiple V, J, D, and C gene segments. Somatic recombination is an orderly process in which D is first rearranged to J (with deletion of the intervening DNA) and then V is rearranged to DJ (with deletion of the intervening DNA) (Fig. 8.2). Approximately 150 V, 30 D, and six functional J genes are known. The V genes are grouped into major families. The VDJ product is joined to the C region at the messenger RNA level as transcribed noncoding intron sequences are spliced out. The VDJ is initially joined to Cμ (and adjacent Cδ) but heavy chain switching, which is a separate gene rearrangement event that occurs later in B-cell development, can result in the VDJ being joined to other C gene segments (γ, α, and ε).
When performing Southern blot analysis of the IgH gene, both joining region (JH) and constant region (Cμ) probes are available. Cμ probes were first available historically, but JH probes are more reliable for detecting IgH gene rearrangements (Fig. 8.3) because the Cμ region is deleted when Ig heavy chain class switching occurs.
Immunoglobulin-κ Light Chain Gene
The Igκ gene at chromosome 2p11 is organized like the IgH gene except that there are no D segments and only a single
C region. However, nearly 100 V and five J segments can recombine to produce diversity. When the Igκ gene rearranges nonproductively, a κ-deleting element, located 3′ to the Cκ region, is responsible for deleting Cκ and sometimes also Jκ (11). This occurs in some B cells expressing Igκ and in all B cells expressing Igλ. For this reason, Jκ probes are most informative when this gene is analyzed by Southern blot analysis.
C region. However, nearly 100 V and five J segments can recombine to produce diversity. When the Igκ gene rearranges nonproductively, a κ-deleting element, located 3′ to the Cκ region, is responsible for deleting Cκ and sometimes also Jκ (11). This occurs in some B cells expressing Igκ and in all B cells expressing Igλ. For this reason, Jκ probes are most informative when this gene is analyzed by Southern blot analysis.
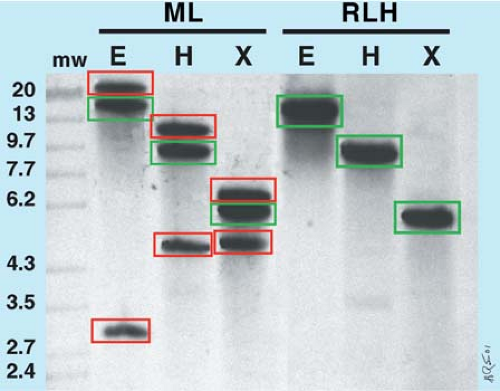 Figure 8.3. Southern blot analysis of immunoglobulin heavy chain (IgH). A radioactively labeled JH probe detects germline bands (green boxes) in B-cell malignant lymphoma (ML) and reactive lymphoid hyperplasia (RLH). Only ML has novel, nongermline, rearranged bands (red boxes), which indicate bi-allelic monoclonal IgH rearrangement. mw, molecular-weight markers in kilobases. Restriction endonucleases: E, EcoRI; H, HindIII; X, XbaI. (Figure courtesy of H. Ratech, M.D.) |
Immunoglobulin-λ Light Chain Gene
The Igλ gene at chromosome 22q11 is organized differently from that of the other Ig genes: in addition to multiple V gene segments, J and C gene segments occur in tandem. Because of polymorphisms in the human population, the number of J and C gene segments can vary between six and nine (15). Cλ probes are available for Southern blot analysis. However, the number of Cλ regions and the polymorphic nature of this region yields a variable number of germline bands, making it difficult to appreciate a true gene rearrangement. For these reasons, the Igλ gene is not usually evaluated by Southern blot analysis.
T-Cell Receptor-β Chain Gene
The TCRβ chain gene at chromosome 7q34 is composed of multiple V gene segments, and two sets of D, J, and C gene segments arranged in tandem. Each set includes one D, six J, and one C gene segment. Following DJ rearrangement, VDJ gene rearrangement can involve either the Jβ1 or Jβ2 gene segments. Southern blot analysis of the TCRβ gene is the most reliable means of assessing T-cell clonality, detecting TCRβ rearrangements in 90% to 95% of T-cell lymphomas and leukemias.
Cβ probes, which were available initially, detect gene rearrangements involving either the Cβ1 or Cβ2 constant regions. The location of an EcoRI restriction enzyme site between Jβ2 and Cβ2 renders rearrangements involving Jβ2 undetectable in EcoRI-digested DNA. Similarly, a Hind III site between Jβ1 and Cβ2 renders rearrangements involving Jβ1 undetectable in Hind III-digested DNA. Jβ probes do not suffer from this limitation; typically, a mixture of Jβ1 and Jβ2 probes is used.
One advantage of using a Cβ probe is that it allows one to estimate the percentage of polyclonal T cells. In EcoRI-digested DNA, the intensity of the 12–kb Cβ1 germline band diminishes proportionally to the percentage of polyclonal T cells in the sample (Fig. 8.4). This is because each polyclonal T cell that has a gene rearrangement involving Cβ1 migrates differently according to a log-linear relationship of molecular mass versus gel distance. In contrast, Cβ2 is not diminished in intensity, in comparison with the germline band, when either Cβ1 or Cβ2 is rearranged because the EcoRI site is 5′ to the Cβ2 allele (1).
T-Cell Receptor-γ Chain Gene
The TCRγ gene at chromosome 7q15 is far simpler than TCRβ, consisting of 11 functional V gene segments that can be subdivided into four families. In addition, J and C segments occur in tandem, with five total J segments (three Jγ 1 and two Jγ2) and two total C segments (Cγ1 and Cγ2). TCRγ gene rearrangements are retained in nearly all normal and neoplastic T cells expressing TCRαβ protein, including most peripheral and cutaneous T-cell lymphomas.
The simplicity of the TCRγ gene makes it better suited for analysis by PCR rather than Southern blot hybridization. Southern blot analysis has a sensitivity of 1% to 5%. The limited number of possible Vγ–Jγ gene rearrangements means that Southern blot analysis will detect discrete bands when numerous polyclonal T cells are present. This results in confusing band patterns when diagnostic biopsy specimens composed of monoclonal and polyclonal T-cell populations are analyzed (16). For these reasons, Southern blot analysis of the TCRγ gene can be problematic for molecular diagnosis.
T-Cell Receptor-α Chain Gene
The TCRα gene at chromosome 14q11 is very large and not suitable for Southern blot analysis. The Jα region is approximately 80 kb, three to four times the size of the largest DNA fragment (20 to 25 kb) routinely studied by Southern blot analysis, and it has approximately 50 Jα gene segments that require multiple probes for analysis (1,2). However, this region can be evaluated in a research environment with the use of pulsed-field gel electrophoresis, which is capable of resolving extremely large DNA fragments.
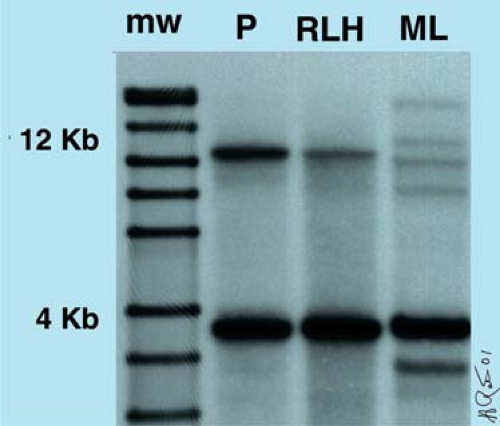 Figure 8.4. Southern blot analysis of T-cell receptor (TCR) β. The DNA was digested with restriction endonuclease EcoRI and a radioactively labeled Cβ probe was used. The intensity of the 12–kb Cβ1 germline band (P) diminishes proportionally to the percentage of polyclonal or monoclonal T cells that have rearranged this DNA region in the test sample. mw, molecular-weight markers in kilobases; P, placenta; RLH, reactive lymphoid hyperplasia; ML, T-cell malignant lymphoma. (Figure courtesy of H. Ratech, M.D.) |
T-Cell Receptor-δ Chain gene
The TCRδ gene is a relatively small gene, located within the TCRα gene at chromosome 14q11, flanked by Vα and Jα gene segments. The TCRδ gene contains approximately five Vδ, two or three Dδ, and three Jδ segments. During normal T-cell development, TCRδ gene rearrangement plays a role in determining whether a T cell expresses the TCRαβ or TCRγ/δ receptor. If the TCRδ gene rearranges successfully, the cell goes on to become a γ/δ T cell. Otherwise, the TCRδ gene is deleted via Vα to Jα gene rearrangement, which deletes all intervening DNA sequences, including the TCRδ gene. TCRδ gene rearrangements can be detected in precursor lymphoblastic lymphoma/leukemia corresponding to the earliest stages of lymphoid differentiation and also in T-cell leukemias and lymphomas that express the TCRγδ receptor (17). Jδ probes are most helpful for Southern blot analysis.
Polymerase Chain Reaction Analysis of Antigen Receptor Genes
Polymerase Chain Reaction Basics
PCR is a versatile molecular technique for assessing clonality, chromosomal translocations, minimal residual disease, gene mutations, and the presence of infectious agents. The lymph
node sample can be fresh, frozen, stored in buffer, or formalin-fixed and routinely processed. The target molecule can be either genomic DNA (standard PCR) or messenger RNA, which, by using reverse transcriptase (RT), is converted to complementary DNA before standard PCR is performed. This is known as reverse transcriptase PCR (RT-PCR). Repetitively altering the reaction temperature during heat denaturation, primer annealing, and extension geometrically amplifies the template DNA sequence 2N times, where N equals the number of cycles (2,3). Multiplex PCR assays also can be set up. A multiplex PCR assay simply combines two or more PCR reactions within the same test tube. Setting up a successful multiplex PCR assay is more complex, because the primers and PCR conditions must be optimized for all the PCR reactions in the tube.
node sample can be fresh, frozen, stored in buffer, or formalin-fixed and routinely processed. The target molecule can be either genomic DNA (standard PCR) or messenger RNA, which, by using reverse transcriptase (RT), is converted to complementary DNA before standard PCR is performed. This is known as reverse transcriptase PCR (RT-PCR). Repetitively altering the reaction temperature during heat denaturation, primer annealing, and extension geometrically amplifies the template DNA sequence 2N times, where N equals the number of cycles (2,3). Multiplex PCR assays also can be set up. A multiplex PCR assay simply combines two or more PCR reactions within the same test tube. Setting up a successful multiplex PCR assay is more complex, because the primers and PCR conditions must be optimized for all the PCR reactions in the tube.
Primer Design
In clinical PCR applications, the DNA target usually spans 2,000 bp or less. In fixed, paraffin-embedded tissues, targets of less than 200 bp are ideal. Prior knowledge of the sequences upstream (5′) and downstream (3′) of the target is required to synthesize short, complementary oligonucleotides (20 to 30 bp), also called primers or amplimers. The primers specifically anneal to the template DNA and provide the initial sequences for Taq DNA polymerase extension. Selecting optimal sequences for both 5′ and 3′ primers often involves compromise; the desire for equivalent guanine and cytosine (G+C) content, which affects melting temperature, must be balanced against the need to avoid complementarity between the 3′ ends, which promotes wasteful primer–dimer formation instead of amplifying the template (18,19,20).
Application to Antigen Receptor Gene Rearrangements
Most DNA-based PCR strategies to detect Ig or TCR gene rearrangements exploit the change in distance between V and J segments that occurs after gene rearrangement (2,3,4). In the germline configuration, V, D, and J gene segments are too widely dispersed for PCR to generate VDJ or VJ products (also known as amplicons) (Fig. 8.1). By contrast, in VDJ or VJ gene rearrangements, the V and J segments are in close proximity and can be amplified by PCR (Fig. 8.2). Upstream and downstream consensus primers are designed to recognize homologous DNA sequences of V genes, such as the framework regions or leader sequence, and the 5′ end of J genes, respectively.
Once amplification is complete, the PCR products must be detected. Traditionally, amplification products were subjected to electrophoresis, during which they separate according to their size (21,22). Monoclonal populations, yielding one or two overrepresented homogeneous PCR species, appear as sharp bands (Fig. 8.5); polyclonal populations yielding many heterogeneous PCR species, each differing in length by a few base pairs, appear as a smear (2,3,4). Alternative gel systems, either single-stranded conformation polymorphism gel electrophoresis or denaturing gradient gel electrophoresis, also can be used (23). In these systems, the PCR product nucleotide sequence determines gel mobility by affecting secondary or tertiary DNA structure.
More recently, fluorescent dyes are being incorporated into primers so that the PCR amplicons are fluorescent (23,24,25,26). Products are then separated by capillary electrophoresis and analyzed by genetic analyzers armed with sophisticated software (e.g. GeneScan) (Fig. 8.6). This new approach has many advantages. Turnaround time is faster; the number of setup steps is reduced, thereby reducing technician error; the detection of fluorescent products is more sensitive than naked-eye examination of a gel; and amplicons can be sized reliably, thereby allowing the comparison of product size over time in minimal residual disease applications (23,24,25,26).
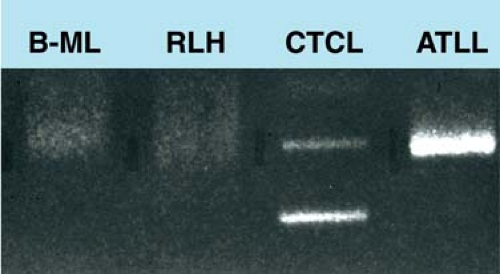 Figure 8.5. Polymerase chain reaction analysis of T-cell receptor (TCR) γ. Polyclonal T cells in B-cell lymphoma (B-ML) and reactive lymphoid hyperplasia (RLH) show smears. Monoclonal T cells in cutaneous T-cell lymphoma (CTCL) and adult T-cell leukemia/lymphoma (ATLL) show one or two sharp bands. Although B-ML contains a clonally rearranged immunoglobulin heavy chain gene, the TCR γ genes in the lymphoma cells remain germline; however, reactive T cells in this sample have numerous different TCR γ gene rearrangements (polyclonal) appearing as a smear. |
False-Positive and False-Negative Results
Because of the high sensitivity of PCR assays, very small monoclonal B- or T-cell populations can be identified in histologically and immunophenotypically benign specimens. These may be viewed as “false-positive” results, although the assay is truly detecting a small monoclonal cell population. However, the clinical and biologic significance of this small clone in the clinicopathologic setting is unknown. In general, when evaluating a histologically equivocal biopsy specimen, a positive PCR result is less persuasive than a positive Southern blot analysis result—because the latter is less sensitive, it detects a larger monoclonal population of cells. One must also avoid the overinterpretation of faint bands or a peak in a background of polyclonal cells seen as an underlying smear or bell curve of peaks. These results are often truly false-positive. Clinicopathologic correlation is always required in the analysis of PCR data.
PCR assays can also yield false-negative results. In the assessment of the IgH gene by PCR, two major causes of false-negative results are possible: (a) consensus V region primers may not anneal with all possible V regions involved in gene rearrangement, and (b) somatic mutations of the immunoglobulin V regions can result in poor annealing of the consensus V primer with the DNA target (2,3). Certain types of B-cell lymphoma (e.g., follicular lymphoma) are much more prone to somatic mutation than other types, meaning that the false-negative rate is also related to lymphoma type (2,19). By using multiple primer sets and analyzing both the IgH and Igκ genes, the false-negative rate can be substantially reduced but usually not eliminated in modern PCR protocols (27,28).
PCR versus Southern Blot to Assess Antigen Receptor Gene Rearrangements
PCR-based methods have replaced Southern blot analysis for the routine assessment of Ig and TCR antigen receptor gene rearrangements (Fig. 8.4). The sensitivity of PCR-based methods for the detection of monoclonal antigen receptor gene rearrangements is approximately 1%. The major advantages of PCR-based methods over Southern blot analysis are that they allow faster turnaround time, are less laborious, can be performed on very small biopsy specimens, and allow assessment of formalin-fixed, paraffin-embedded tissue specimens. PCR-based assays, unlike Southern blot analysis, are also more specific in the sense that they only analyze lymphoid cells; Southern blot analysis analyzes all cells in a specimen (e.g., stromal cells,
histiocytes, etc.). This results in PCR-based assays being more sensitive than Southern blot analysis in the evaluation of specimens with a mixture of cell types. Most laboratories now use PCR-based methods initially or exclusively to assess for antigen receptor gene rearrangements. As discussed earlier, the major disadvantage of PCR-based assays is that that can yield false negative results. However, by assessing multiple antigen receptor genes for clonality (e.g., IgH and Igκ or TCRγ and TCRβ), false-negative results are infrequent. Although the most recent protocols using multiplex assays and the analysis of multiple antigen receptor genes have decreased the percentage of false-negative results, they still occur in a subset of cases (27,28). Thus, Southern blot analysis of the antigen receptor genes is still considered by some molecular pathologists to be the most reliable test to assess clonality (Table 8.1).
histiocytes, etc.). This results in PCR-based assays being more sensitive than Southern blot analysis in the evaluation of specimens with a mixture of cell types. Most laboratories now use PCR-based methods initially or exclusively to assess for antigen receptor gene rearrangements. As discussed earlier, the major disadvantage of PCR-based assays is that that can yield false negative results. However, by assessing multiple antigen receptor genes for clonality (e.g., IgH and Igκ or TCRγ and TCRβ), false-negative results are infrequent. Although the most recent protocols using multiplex assays and the analysis of multiple antigen receptor genes have decreased the percentage of false-negative results, they still occur in a subset of cases (27,28). Thus, Southern blot analysis of the antigen receptor genes is still considered by some molecular pathologists to be the most reliable test to assess clonality (Table 8.1).
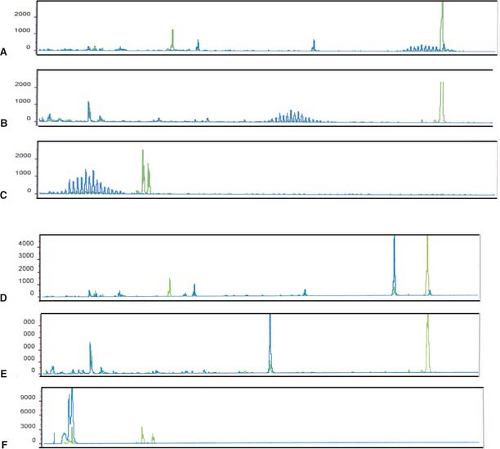 Figure 8.6. Two clinical specimens analyzed for the presence of the IgH gene rearrangements using three sets of fluorescently labeled primers followed by capillary electrophoresis and GeneScan analysis. In the first specimen (A–C), a polyclonal population of cells is present shown by the bell shaped curves (blue). In the second specimen (D–F), a monoclonal population of cells is present shown as a discrete peak (blue). The control in each lane is green. In each primer set, one primer is designed to anneal to the V framework region 1 (A,D), V framework region 2 (B, E), and V framework region 3 (C, F), respectively and the other primer is a mixture that anneals with the J region. |
Molecular Diagnosis of Chromosomal Translocations
Oncogene activation is commonly involved in the pathogenesis of malignant lymphomas. Although oncogenes can be activated via a number of mechanisms, the most common mechanism in lymphomas is via chromosomal translocation (5,29,30,31). Two general types of translocation occur. In the first type, the translocation juxtaposes an oncogene with either an Ig or TCR gene; the reading frame of the oncogene is not disrupted. Thus, the translocation results in overexpression of qualitatively normal oncogenic protein. Examples of this type include IgH juxtaposed with myc in Burkitt lymphoma (Fig. 8.5) and cyclin D1 in mantle cell lymphoma (Fig. 8.6). Mistakes in physiologic VDJ recombination or IgH heavy chain class switching are likely to be involved in generating this type of translocation. The second type of translocation does not involve antigen receptor gene loci. Instead, a gene on each partner chromosome is disrupted; these genes are then recombined to create a novel gene. Examples of this type include alk translocations in anaplastic large-cell lymphoma and the t(11;18)(q21;q21) in extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT). The mechanisms involved in the pathogenesis of this type of translocation are poorly understood.
Chromosomal translocations can be detected by conventional cytogenetics, FISH, Southern blot analysis, and PCR-based methods. The best method to detect a chromosomal translocation depends on the type of sample available, the anatomy of the genes involved, the number and locations of the breakpoints, and the goals of the analysis (e.g., initial diagnosis versus detection of minimal residual disease).
TABLE 8.1 DETECTION OF ANTIGEN RECEPTOR GENE REARRANGEMENTS | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Conventional Cytogenetics
Virtually all of the known chromosomal translocations in malignant lymphomas were first detected by conventional cytogenetic analysis, providing the preliminary data leading to the cloning of the oncogenes involved in these translocations. This is because conventional cytogenetic analysis allows for global chromosome analysis (32). In other words, conventional cytogenetic analysis allows one to observe grossly visible abnormalities involving the 46 chromosomes. If a specific oncogene involves multiple partner genes, or if breakpoints involving a chromosomal translocation are highly variable, conventional cytogenetics can detect these variations. By contrast, FISH and other molecular methods are much more focused, assessing for a specific abnormality. These methods also do not allow assessment of all partner genes, and some molecular methods, particularly PCR approaches, are compromised by multiple or widely spaced breakpoints.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


