Chapter 10 Metabolism and pharmacokinetic optimization strategies in drug discovery
Introduction
Optimization of drug metabolic and pharmacokinetic properties is an integral component of the modern drug discovery process. The objective of the drug metabolism and pharmacokinetics (DMPK) discipline in drug discovery is to aid design and selection of candidate drugs with properties that yield the required efficacy and safety for effective clinical use. The roles of DMPK at the various stages of drug discovery are summarized in Table 10.1. DMPK in vitro and in vivo information are used throughout the drug discovery process to facilitate target validation and safety assessment, and to guide the conversion of early screening hits and leads into drug candidates. Indeed, the frontloading of DMPK in drug discovery has resulted in a reduction of drug attrition rate due to undesirable DMPK properties from approximately 40% in 1990 to 10% in 2000 (Kola and Landis, 2004).
Table 10.1 Roles of drug metabolism and pharmacokinetics (DMPK) in various phases of drug discovery
Optimization of DMPK properties
Optimization principles are described for six key DMPK areas:
• Absorption and bioavailability
• Avoiding PK-based drug–drug interactions
• Achieving/avoiding CNS exposure
• Role of metabolite identification:


Absorption and oral bioavailability
Introduction
As the preferred route of administration for most indications is oral, it is important to characterize oral bioavailability (F) of a compound during drug discovery. In addition, F must be optimized, as a low F is often associated with poor and variable exposure and lack of efficacy. F is defined as the percentage of dosed drug that reaches the systemic circulation compared to the IV route. As shown below, it can be considered to be dependent on three serial steps: the fraction of dosed drug absorbed (fa), the fraction escaping intestinal metabolism (fg) and the fraction extracted by the liver as it passes from the portal vein to the systemic circulation (fh) (see Rowland and Tozer, 1989):
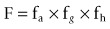
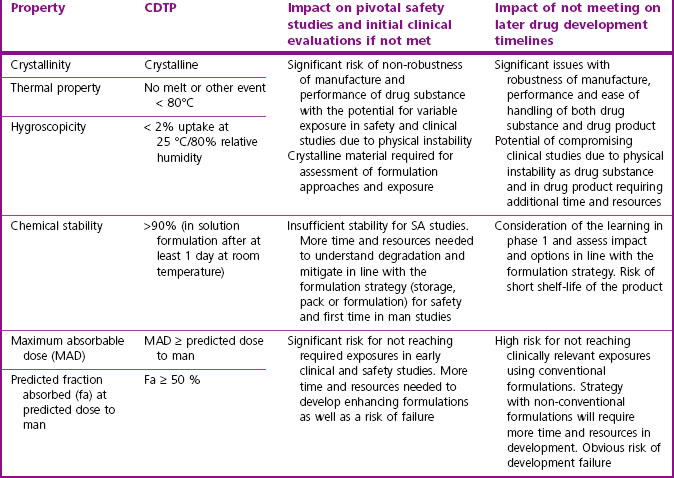
In humans, the combinations of high to low solubility and permeability have led compounds to be characterized according to the Biopharmaceutical Classification System (Amidon et al., 1995). Class 1 compounds, with high solubility and permeability, generally have very good absorption properties. Those in Class 4, with poor solubility and permeability, are likely to present significant formulation challenges and/or variable and poor exposure. It can be important to characterize the maximum absorbable dose (MAD) of a compound relative to its predicted therapeutic dose, as this will determine the risk of being able to deliver an efficacious dose to humans and guide whether high exposure in safety studies is achievable.
Table 10.2 gives guidance on acceptable pharmaceutical properties for typical oral drug candidates.
Tactics
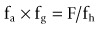
When maximizing the chances of good absorption, the starting point is to ensure that the physicochemical properties of the compound/series are in the optimal space as described by Lipinski and others (Lipinski et al., 1997; Wenlock et al., 2003; Johnson et al., 2009; Waring, 2009). Generally, this requires minimizing the number of H-bond donors and acceptors, restricting lipophilicity in the range LogD7.4 0 to 3, and limiting molecular weight to <500. Both the solubility and passive permeability of a compound should be assessed prior to any in vivo study. Predictive models for these should be assessed for suitability in each project and considered in compound design if either is an issue for a chemical series.
The primary in vitro tool for assessing absorption is the cell based Caco-2 permeability assay, although MDCK-MDR1, PAMPA (parallel artificial membrane permeability assay), or in silico predictions may also provide valuable information about efflux transporter risk and permeability. This permeability assessment, in combination with a solubility measurement (ideally using crystalline material), is used to estimate fa using commercially available modelling tools like GastroPlus (www.simulations-plus.com) or SIMCYP (www.simcyp.com). For actively transported (efflux or uptake) compounds, bi-directional permeability assays can be used as a guide to possible in vivo effects. However, it should be noted that it is often difficult to extrapolate the results from these in vitro transport assays to accurately quantify effects in vivo. Because of its reasonable throughput, the Caco-2 assay can be positioned as an early screen if absorption or permeability/efflux is found to be an issue in the project. If further evaluation of absorption or efflux is warranted, it is possible to use more physiological models such as sections of intestinal tissue in an Ussing Chamber (Ungell et al., 1998), or transfected cell lines over-expressing particular efflux transporters (e.g. P-gp in the MDCK-MDR1 cell line). The Ussing Chamber technique can help in understanding cross-species differences and, because the tissue used is enzymatically competent (metabolic and transporters), the output represents the product of fa and fg.
Compounds with low hepatic clearance in the rat, good solubility and high effective permeability should exhibit good oral absorption and bioavailability in that species. However, if this is not the case, the troubleshooting decision tree in Figure 10.1 can be used to help determine the cause(s) of poor absorption, identify assays to aid in optimizing compound design and understand if the compound is of sufficient quality to progress in the value chain, despite its non-optimal absorption properties.
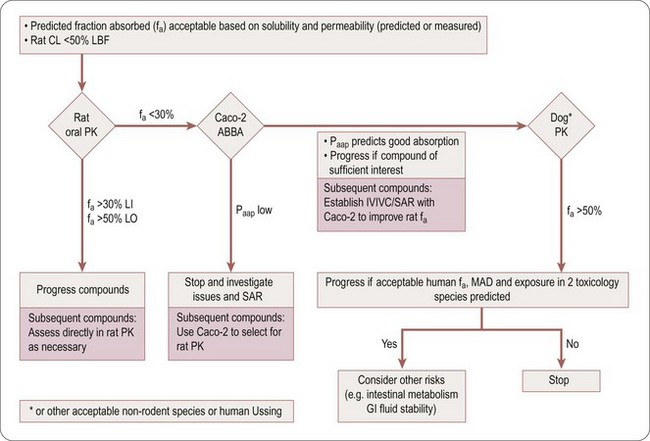
Table 10.3 is an aid to selecting the assays and techniques to use in the decision tree (Figure 10.1), to explain potential issues and risks, and the parameter (fa, fg and/or fh) the assays impact on.
Table 10.3 Assays and techniques used when troubleshooting absorption
| Assay/technique | Potential issue/risk addressed | Impacted |
|---|---|---|
| Intestinal microsomes or S9 fraction | Assess cross species differences in intestinal metabolism. Nature and source of metabolites can give key information about potential for gut metabolism, as CYP3A and UGTs account for most gut metabolites | fg |
| Absorption profiling on Caco-2 cells | Varying apical to basolateral pH gradient Concentration dependency Use of proteins (e.g. BSA) at various percentages in apical chamber Use of efflux transporter inhibitors | fa |
| PAMPA | Passive permeability | fa |
| High dose PK studies | Saturation of efflux or metabolism | fa, fg, fh |
| Ussing chamber technique | Effective permeability (including transporters) Intestinal tissue metabolism | fa, fg |
| GI stability test | Degradation of drug in stomach or intestinal lumen is possible explanation (usually the case if predicted F much greater than measured F) | fa |
| Human metabolic phenotyping | Gut metabolism | fg, fh |
| In situ/in vivo portal vein cannulation preparation | Determine amount of drug and metabolites passing through intestine | fa, fg |
| SIMCYP; GastroPlus | Predict absorption rate and extent (software methods) | fa |
| Transfected cell lines; vesicles expressing specific transporters; Caco-2 efflux assay with and without specific transporter inhibitors | Determine involvement of specific efflux transporters | fa, fh |
| Knock-out (KO) animals; chemical KO with inhibitors | Determine involvement of specific efflux transporters | fa, fg, fh |
Cautions
• As scaling factors for intestinal microsomes or other subcellular intestinal fractions are currently not available, it is difficult to make an accurate quantitative assessment of the contribution of intestinal metabolism to in vivo fa. However, attempts have been made to use CLint from human liver microsomes or S9 fraction to estimate the relative contributions of fa and fg to F (Gertz et al., 2010).
• It is often very difficult to pinpoint why compounds have poor absorption characteristics and, therefore, to resolve this design issue. Thus, it is often reasonable to prioritize series with good fa x fg, in early discovery even if other properties (e.g. potency, clearance) are less attractive.
• Typically, oral doses are formulated as suspensions which, on many occasions, may be derived from amorphous material. However, it is important to assess absorption periodically using crystalline material, as physical form may have substantial effects on absorption profiles.
• Particularly for compounds likely to proceed into development, it is important to determine the effect of the polymorphic solid states on absorption. It is also important to ensure that the formulations used are discussed with Pharmaceutical Development (see Chapter 16), and are appropriate for safety and early clinical development studies.
• If in vitro and in vivo (rat and dog) assessments of fa do not agree, the risk of an inaccurate estimate of absorption in man will increase. Sometimes other species have been investigated to mitigate this risk, but we caution that there can, for example, be marked discrepancies in fa x fg between cynomolgus monkeys and humans (Takahashi et al., 2009).
Avoidance of PK based drug–drug interactions
Introduction
• Competitive (reversible) cytochrome P450 (CYP) inhibition
• Mechanism based/time dependent CYP inhibition
• Uptake and efflux transporter inhibition
CYP, notably CYP3A4 and CYP2D6, based DDI is the most important and most common, and may occur in the liver or intestine. Transporter based DDI is mainly related to renal clearance, although specific issues can arise with CNS compounds and hepatic uptake of statins. The science and regulatory guidance to support risk assessment of CYP-based DDI are well established, but is less advanced for transporter-related issues (Bjornsson et al., 2003; US Food and Drug Administration, 1997, 2006; Huang et al., 2008; International Transporter Consortium, 2010).
Tactics
Competitive (reversible) CYP inhibition
Two main types of CYP inhibition assays with different capabilities are in general use. Fluorescence based assays are relatively cheap and have enhanced throughput, but in a small but significant number of cases can lead to misrepresenting DDI risk (Bell et al., 2008). They are best used for initial profiling of large numbers of compounds, with the data being acceptable in the early phase such as lead generation. LCMS based assays are more expensive, have lower throughput, but are more predictive. They should be used in optimization cycles once CYP inhibition issues have been identified, and for generating compound profiles during more advanced project phases.
Reduction of CYP inhibition potential is facilitated by the fact that strong QSAR relationships are often obtained. Various computational models that allow prediction of CYP DDI risk are available within most drug development companies. It is well established that lipophilicity, aromaticity and charge type are major drivers for inhibiting various CYP enzymes (Gleeson et al., 2007).
The risk of DDIs based on Phase 2 metabolism (e.g. glucuronidation and sulphation) is usually small, resulting in less than a two-fold increase in area under the concentration versus time curve (AUC), and they are rarely observed, possibly due in part to the nature of the enzymatic reaction (high Vmax and moderate to high Km values). Such DDIs are not generally evaluated in lead optimization (Williams et al., 2004). Evidence suggesting the need to do so at this stage should prompt re-evaluation of the risk.
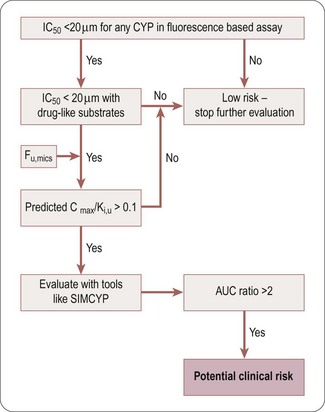
The decision tree in Figure 10.2 can be used to assess the potential DDI risk of a competitive CYP inhibitor. Hits identified in a fluorescence-based assay should be confirmed with an LCMS-based assay using druglike substrates as probes for the different CYP isoforms. Although the ratio Cmax/Ki,u can be used to obtain a preliminary estimate of the DDI risk, more accurate evaluation should be conducted using PBPK modelling (e.g. SIMCYP platform) to predict the potential clinical risk (expanded below in ‘Prediction of DDI risk’ ”).
Mechanism-based/time-dependent CYP inhibition
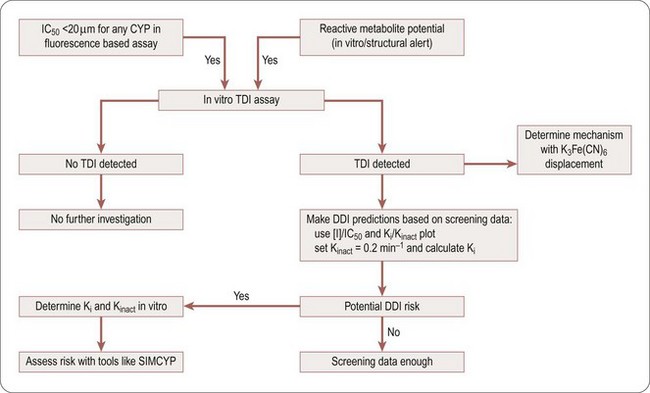
The inhibition of CYP enzymes may be irreversible (due to irreversible or covalent binding to the prosthetic haem or the enzyme) or quasi-irreversible (due to the formation of transient complexes with the iron of the haem prosthetic group). Time-dependent inhibition (TDI) methods can be used to determine this (Riley et al., 2007; Fowler and Zhang, 2008). During early phases of drug discovery, a medium throughput screening assay can be used to screen for TDI. However, for selecting candidates at later phases, the method employed should provide accurate determination of Kinact and Ki to properly evaluate the DDI risks of compounds in which preliminary screening indicated the potential for TDI. A positive TDI finding also suggests that the compound or its metabolites may be reactive, and further evaluation should be conducted as specified by reactive metabolite strategies.
A decision tree to help evaluate the potential DDI risk of a TDI CYP inhibitor is shown in Figure 10.3. If a compound is found to be at risk for CYP inhibition (IC50 <20 µM) or is flagged to have a potential liability for reactive metabolites, it should be screened for TDI. If it is found to have potential TDI risk based on screening data, Ki and Kinact values should be generated to help accurately predict the risk using prediction tools like SIMCYP.
Uptake and efflux transporter inhibition
Uptake transporter inhibition assays are emerging as being of real value (Ward, 2008), but they are highly dependent on chemotype (e.g. acids being primarily transported by organic anion transporters (OATs)) and likely co-medications (e.g. OCT2 and metformin). Acids and zwitterions should be assessed for inhibition of OATP1B1 during drug discovery. Other inhibition assays (OAT1, OAT3, OATP1B1, OCT1 and OCT2) should be used on a case-by-case basis.
CYP induction mediated risk for DDI
Induction of specific CYP enzymes may not only change a drug’s metabolic profile but also have toxicological consequences as CYP enzymes are also involved in the metabolism and synthesis of important endogenous compounds. Although close collaboration with Safety Assessment (see Chapter 15) is needed to evaluate the full impact of CYP induction, it is DMPK’s primary responsibility to predict the DDI potential caused by CYP induction in man. This should be done during optimization with the use of HepaRG cells which are a good surrogate of primary human hepatocytes for AhR-mediated CYP1A induction and PXR- and CAR-mediated CYP3A4 and CYP2B6 induction. If higher throughput is needed during the optimization phase, the PXR reporter gene assay may be used to minimize PXR-dependent CYP3A4 induction liability.
Prediction of DDI risk
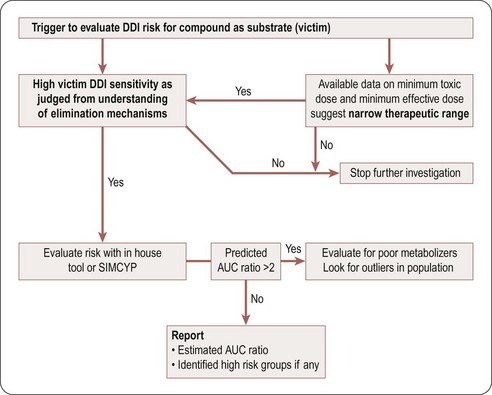
A compound is likely to be a victim of clinically relevant DDI with a co-medication if it has a narrow therapeutic window and is a sensitive substrate to an inhibited enzyme/transporter, as indicated by high values of fraction metabolized (fm), fraction of CYP metabolized (fm,CYP), and fraction unbound (fu), and by plasma and hepatic extraction. This risk can be reduced by ensuring that the clearance mechanism in question represents <50% of total clearance (resulting in less than a two-fold change in the AUC of the victim compound). At candidate drug nomination, software tools such as SIMCYP could be used to evaluate the impact of known inhibitors or inducers on the relevant candidate compounds if their clearance is driven mainly by a single enzyme or transporter. Figure 10.4 is a decision tree to help in assessing the potential risk that a compound will be a DDI victim. A compound is deemed to have this potential risk if it has a narrow therapeutic window or is cleared predominantly by a single CYP enzyme. A simple Excel based DDI template or SIMCYP should be used to estimate the risk. If the CYP enzyme involved is polymorphic, evaluate the impact of polymorphism to identify high-risk populations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


