Metabolic Effects of Tumours
This chapter looks at how clinical biochemistry tests can be used to diagnose and monitor patients with malignant disease. Malignant disease is one of the world’s major ‘killers’, and it is therefore important to know the basic principles of biochemical diagnosis and monitoring in such patients.
Neoplastic cells of differentiated tissues sometimes synthesize enough compounds not normally thought of as coming from that tissue to be detectable in body fluids. These substances fall into two principal groups:
those that alter metabolism and thus may produce clinical effects, some of which are hormonal syndromes,
those that, although biologically inactive, may be analytically detectable in body fluids; these are sometimes used as tumour markers.
THE DIFFUSE ENDOCRINE SYSTEM
Certain rare syndromes with endocrine or neurotransmitter properties are associated with neoplasia. These cells may share common cytological characteristics, originating in the embryonic ectoblast. Staining techniques have shown their ability for amine precursor uptake and decarboxylation (APUD) with the production of amines. Tumours of these cells have therefore been called APUDomas (although this older classification has been challenged). Some secrete physiologically active amines, whereas others, such as the pituitary and parathyroid glands and the calcitonin-producing C cells of the thyroid, secrete peptide hormones. Many occur in the gastrointestinal tract and pancreas. Secreting tumours of tissues of the sympathetic nervous system and the amine-secreting carcinoid tumours are discussed in this chapter.
CATECHOLAMINES
The sympathetic nervous tissue comprising the adrenal medulla and sympathetic ganglia is derived from the embryonic neural crest and is composed of two types of cells, the chromaffin cells and the nerve cells, both of which can synthesize active catecholamines (dihydroxylated phenolic amines). Adrenaline (epinephrine) is almost exclusively a product of the adrenal medulla, whereas noradrenaline (norepinephrine) is predominantly formed at sympathetic nerve endings.
CASE 1
A 34-year-old man was referred to the hypertension clinic because of uncontrolled hypertension. His blood pressure in the clinic was 186/104 mmHg, despite treatment with bendroflumethiazide, enalapril and felodipine. Some biochemistry tests were requested, which gave the following results:
Plasma
Sodium 138 mmol/L (135-145)
Potassium 4.2 mmol/L (3.5-5.0)
Urea 5.7 mmol/L (2.5-7.0)
Creatinine 90 µmol/L (70-110)
Urine
Normetanephrine 23.1µmol/24 h (normally < 2.0 µmol/24 h)
Metanephrine 17.3 µmol/24 h (normally < 1.5 µmol/24 h)
DISCUSSION
Phaeochromocytoma is the likely diagnosis, given the raised urinary metanephrine concentrations. This was confirmed by further tests, including a radioisotope metaiodobenzylguanidine (MIBG) scan and an adrenal MRI scan. The MEN syndrome needs to be excluded, given the known association with phaeochromocytoma. It is often wise to seek a secondary cause of hypertension in young individuals with hypertension, particularly those refractory to treatment.
Metabolism of catecholamines
Adrenaline and noradrenaline are formed from the amine precursor tyrosine via dihydroxyphenylalanine and dihydroxyphenylethylamine (dopamine) and bear the catechol (3,4-dihydroxyphenyl)-moiety. Adrenaline and noradrenaline are both metabolized to the inactive 4-hydroxy-3-methoxymandelic acid (HMMA), incorrectly called vanillyl mandelic acid (VMA), by similar pathways in which metadrenaline and normetadrenaline, respectively, are intermediates (Fig. 24.1). Adrenaline, noradrenaline and their metabolites can be measured in urine.
Action of catecholamines
Noradrenaline causes generalized vasoconstriction, with hypertension and pallor. Adrenaline dilates blood vessels in muscles, with variable effects on blood pressure and pulse rate. Adrenaline may also cause hyperglycaemia due to stimulation of glycogenolysis and other anti-insulin effects.
Catecholamine-secreting tumours
Phaeochromocytomas
Phaeochromocytomas present mainly in adults and occur in chromaffin tissue. About 10 per cent are outside the adrenal medulla, 10 per cent are bilateral and 10 per cent are malignant. They are associated with the multiple endocrine neoplasia (MEN) syndrome, neurofibromatosis and Von Hippel-Lindau disease although many are isolated tumours. The symptoms and signs can be related to very high plasma concentrations of catecholamines.
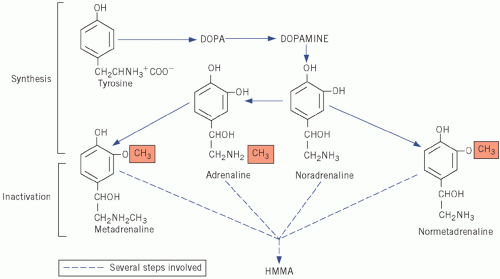 Figure 24.1 Synthesis and metabolism of catecholamines. DOPA, dihydroxyphenylalanine; HMMA, 4-hydroxy-3-methoxymandelic acid. |
The hypertension associated with phaeochromocytoma is potentially curable by surgery (Fig. 24.2). Its presence should be excluded, particularly if a young adult presents with paroxysmal or persistent hypertension for no apparent reason or a patient presents with refractory hypertension or shows the classic triad of sweating, headaches and tachycardia associated with hypertension. Hyperglycaemia and, if the plasma glucose concentration is high enough, glycosuria may occur during the attack.
Diagnosis of phaechromocytomas
The biochemical diagnosis of these tumours can be made by measuring the daily urinary excretion of catecholamines (adrenaline, noradrenaline and dopamine) or HMMA, the major catabolic product of catecholamines. Extra-adrenal tumours may only secrete dopamine. Metanephrines (which are also metabolites of catecholamines and include metadrenaline, normetadrenaline and methoxytyramine) are preferable because they are more sensitive than catecholamines and can also be measured in urine and their concentration may be raised in some phaeochromocytomas when urinary catecholamines are normal. A slightly increased excretion of urinary catecholamines can be found in cases of essential
hypertension. Various dietary components and drugs affect some of the analytical methods, and therefore it is important to consult your laboratory before starting a urine collection. Labetalol, paracetamol, α-methyldopa and Sinemet (combined levodopa and carbidopa) may interfere with some of these assays.
hypertension. Various dietary components and drugs affect some of the analytical methods, and therefore it is important to consult your laboratory before starting a urine collection. Labetalol, paracetamol, α-methyldopa and Sinemet (combined levodopa and carbidopa) may interfere with some of these assays.
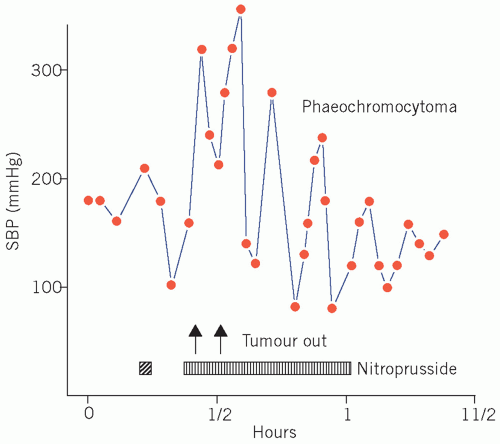 Figure 24.2 Showing large fluctuations in systolic blood pressure during manipulation of a phaeochromocytoma during its surgical removal. The blood pressure was treated with nitroprusside infusion. Adapted with kind permission from Kinirons M and Ellis H. French’s Index of Differential Diagnosis, 15th edition. London: Hodder Arnold, 2011. |
In some cases, particularly if the tumours are small or hypertension is paroxysmal, metabolite excretion in a 24-h urine collection may be misleading. If there is a high index of suspicion, it may be useful to repeat this estimation on three consecutive days. Raised plasma metanephrine concentrations have nearly 100 per cent sensitivity for the diagnosis of phaeochromocytoma and may be particularly helpful if the patient is unable to give a reliable urine sample. However, the concentration of plasma metanephrines may also be raised in renal failure.
The clonidine or pentolinium suppression tests may have a place in the investigation if urinary catecholamines are positive or equivocal and imaging studies are negative. In normal individuals, concentration of urinary and plasma catecholamines should decrease after the administration of the suppressing agent, unlike the situation in a phaeochromocytoma, in which there is autonomous catecholamine secretion.
Imaging studies include abdominal computerized tomography, magnetic resonance imaging (MRI) and metaiodobenzylguanidine (MIBG) uptake scan. The MIBG is taken up specifically by the phaeochromocytoma tissue. The estimation of plasma catecholamine concentrations in samples obtained by selective venous catheterization may help to localize the tumour before surgery.
Neuroblastomas
Neuroblastomas are very malignant tumours of the sympathetic nervous tissue and usually occur in children. About 40 per cent occur in the adrenal medulla, and about 60 per cent are extra-adrenal.
The plasma catecholamine concentrations may be as high as, or higher than, those in patients with phaeochromocytomas. Some neuroblastomas secrete dopamine and its metabolite homovanillic acid (HVA) whose concentration is raised in the urine and may be used as a tumour marker.
THE CARCINOID SYNDROME
Normal metabolism of 5-hydroxytryptamine
Some cells are called argentaffin because they reduce, and therefore stain with, silver salts. These are normally found in tissues derived from the embryonic gut and
are most abundant in the ileum and appendix, but they are also found in the pancreas, stomach and rectum. They synthesize the biologically active amine 5-hydroxytryptamine (5-HT; serotonin) from the amine precursor tryptophan, the intermediate product being 5-hydroxytryptophan (5-HTP). 5-Hydroxytryptophan is inactivated by deamination and oxidation by monoamine oxidases to 5-hydroxyindole acetic acid (5-HIAA) (Fig. 24.3).
are most abundant in the ileum and appendix, but they are also found in the pancreas, stomach and rectum. They synthesize the biologically active amine 5-hydroxytryptamine (5-HT; serotonin) from the amine precursor tryptophan, the intermediate product being 5-hydroxytryptophan (5-HTP). 5-Hydroxytryptophan is inactivated by deamination and oxidation by monoamine oxidases to 5-hydroxyindole acetic acid (5-HIAA) (Fig. 24.3).
CASE 2
A 56-year-old man attended the gastroenterology department because of profuse diarrhoea. He also mentioned that he had felt increasingly breathless, and he looked flushed. A number of investigations were requested, but it was a 24-h urinary 5-hydroxyindole acetic acid (5-HIAA) concentration of 544 µmol/day (less than 25) that suggested the diagnosis.
DISCUSSION
The patient had considerably raised urinary 5-HIAA concentration and symptoms suggestive of carcinoid syndrome. Note the flushing and diarrhoea as well as the breathlessness, probably due to bronchospasm induced by vasoactive and bronchoactive amines. The patient was subsequently found to have carcinoid syndrome involving a tumour of the ileum that had metastasized to the liver. Urine 5-HIAA can be used to monitor disease assuming that a change in management would improve clinical prognosis.
The oxidases and aromatic amino acid decarboxylases are present in other tissues as well as in argentaffin cells; 5-HIAA is usually the main urinary excretion product of argentaffin cells. Argentaffin cells may also excrete the peptide substance P, an excess of which causes flushing, tachycardia, increased bowel motility and hypotension.
Causes of the carcinoid syndrome
The carcinoid syndrome is usually associated with abnormally high concentrations of plasma 5-HT. Ileal and appendiceal tumours only produce the typical clinical syndrome when they have metastasized, usually to the liver. The products of intestinal tumours are inactivated in the liver, but those from other sites, such as the bronchus, are released directly into the systemic circulation in an active form and may therefore cause symptoms.
Only one enzyme, tryptophan-5-hydroxylase, is needed to convert tryptophan to 5-HTP (see Fig. 24.3), and its derepression in malignant syndromes leads to excessive synthesis of 5-HTP. The decarboxylase and monoamine oxidases are present in normal non-argentaffin tissues, so 5-HTP accumulates.
The symptoms and signs of the carcinoid syndrome include:
diarrhoea, which may be severe enough to cause the malabsorption syndrome,
flushing,
bronchospasm and right-sided fibrotic lesions of the heart, such as tricuspid incompetence and pulmonary stenosis; the heart lesions do not occur if the primary tumour is in the bronchus.
The symptoms and signs are more likely to be due to the presence of substance P than of 5-HT. Histamine has also been suggested as a cause of the flushing. A pellagra-type syndrome may occasionally develop, because tryptophan is diverted from nicotinamide to 5-HT synthesis. A carcinoid crisis occurs when the tumour releases large amounts of vasoactive substances into the circulation.
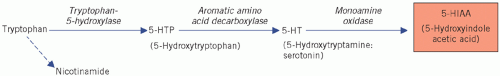 Figure 24.3 Metabolism of tryptophan. |
Diagnosis of the carcinoid syndrome
Urinary 5-HIAA secretion is usually very high in the carcinoid syndrome. A daily excretion of more than 25 µmol is elevated, and greater than 100 µmol indicates carcinoid syndrome if plums, pineapples, kiwi fruit, avocados, tomatoes, walnuts and bananas (which are high in hydroxyindoles) have been excluded from the diet for about 24 h before the urinary collection is started. Elevated urinary 5-HIAA has also been reported in association with small bowel disease and intestinal obstruction, for example coeliac disease and sprue, but usually concentrations are not as high as in carcinoid syndrome.
In very rare cases, usually of bronchial or gastric tumours, the argentaffin cells lack aromatic amino acid decarboxylase. In such cases, despite increased secretion of 5-HTP, urinary 5-HIAA excretion may not be increased to diagnostic levels. If there is a strong clinical suspicion of the carcinoid syndrome, despite the finding of normal 5-HIAA excretion, estimation of total 5-hydroxyindole (5-HTP, 5-HT and 5-HIAA) excretion may be indicated. In some cases, plasma 5-HT or its metabolites may be measured.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


