Membranes and Distribution
OBJECTIVES
The reader will be able to:
- Define the following terms: hydrophilic, hydrophobic, lipophilic, lipophobic, active transport, paracellular transport, passive facilitated transport, permeability, transcellular transport, and transporter.
- List two examples of transporters involved in systemic absorption after oral administration and three involved in the distribution of drugs into and out of tissues, including eliminating organs.
- Define the following terms:
- Perfusion limitation in distribution
- Permeability limitation in distribution
- Tissue-to-blood equilibrium distribution ratio
- Fraction unbound
- Plasma protein binding
- Perfusion limitation in distribution
- Distinguish between perfusion rate-limited and permeability rate-limited passage of drugs through membranes.
- Describe the role of pH in the movement of drugs through membranes.
- Describe the consequences of the reversible nature of movement of drugs through membranes.
- Determine the plasma concentration, the amount of drug in the body, and the apparent volume of distribution at distribution equilibrium when any two of these values are known.
- Describe the effects of perfusion limitation, permeability limitation, and the tissue-to-blood equilibrium distribution ratio on the time required for drug to distribute into and out of tissues.
- Ascertain whether, for a given amount of drug in the body at distribution equilibrium, the unbound plasma concentration is likely to be sensitive to variation in plasma protein binding when the volume of distribution is known.
- From knowledge of the volume of distribution and the fraction unbound for a drug bound only to albumin in plasma, calculate the fraction of drug in the body at equilibrium that is:
- Unbound
- In the extracellular fluids
- Outside the extracellular fluids
- Bound to plasma proteins
- Bound to plasma proteins in the extracellular fluids
- Bound intracellularly (in or on tissue cells)
- Unbound
- Anticipate the effect of altered plasma protein binding on the half-life of a drug, bound to albumin and with a volume of distribution less than 15 L.
 n the last chapter, emphasis was placed on general input–exposure relationships after a single intravenous (i.v.) bolus. We now examine the role and function of membranes primarily in the context of determinants of drug distribution, but the principles apply as well to drug elimination (Chapter 5, Elimination) and drug absorption (Chapter 7, Absorption). Drugs must also pass through membranes to reach the site of action.
n the last chapter, emphasis was placed on general input–exposure relationships after a single intravenous (i.v.) bolus. We now examine the role and function of membranes primarily in the context of determinants of drug distribution, but the principles apply as well to drug elimination (Chapter 5, Elimination) and drug absorption (Chapter 7, Absorption). Drugs must also pass through membranes to reach the site of action.
This chapter also explores the process of distribution itself and its role in clinical pharmacokinetics from a physiologic point of view. The chapter begins with kinetic considerations and ends with equilibrium concepts involved in drug distribution, the reversible transfer of drug from one location to another within the body.
Before examining membranes as a determinant of drug absorption and disposition, a few terms commonly applied to the physicochemical properties of drugs need to be defined. Hydrophilic and hydrophobic are adjectives that refer to water (hydro-) loving (-philic) and fearing (-phobic) properties. Similarly, lipophilic and lipophobic are adjectives that relate to lipid (lipo-) loving and fearing properties. In general, the terms hydrophilic and lipophobic are interchangeable. When referring to substances, both these terms imply that the substances are soluble in water, but very poorly soluble in nonpolar lipids. Similarly, the terms lipophilic and hydrophobic are interchangeable and refer to substances that are soluble in lipids, but very poorly soluble in water. A common measure of lipophilicity of a substance is its partitioning between n-octanol (an organic solvent with hydrogen bonding properties aimed at mimicking the physicochemical properties of tissue membranes) and water. The higher the partition coefficient, ratio of drug concentrations at equilibrium (approximated by the ratio of solubilities) in the two phases, the greater is the lipophilicity. Many drugs contain both hydrophobic and hydrophilic groups and vary in their solubilities in water and n-octanol. Some compounds, such as alcohol, are readily soluble in both water and n-octanol or other lipid-like solvents. Others, such as digoxin, are poorly soluble in both water and lipids. Thus, both solubility and partition coefficient are important in drug absorption and disposition.
MEMBRANES
Movement through membranes is known as drug transport, a term that is often used more specifically to describe the processes and transport systems (transporters) that facilitate movement across membranes. Understanding how physicochemical, anatomic, and physiologic factors determine the rapidity of drug transport is a prerequisite to an appreciation of factors controlling the pharmacokinetics and pharmacodynamics of drugs.
Cellular membranes are composed of an inner, predominantly lipoidal, matrix covered on each surface by either a continuous layer or a lattice of protein (Fig. 4-1, upper section of drawing). The hydrophobic portions of the lipid molecules are oriented toward the center of the membrane and the outer hydrophilic regions face the surrounding aqueous environment. For some drugs, facilitative mechanisms are embedded in the protein lattice. Narrow aqueous-filled channels exist between some cells (e.g., in most blood capillary membranes, the glomerulus of the kidney, and intestinal epithelia).
The transport of drugs is often viewed as movement across a series of membranes and spaces, which, in aggregate, serve as a “functional” macroscopic unit. The cells and interstitial spaces that lie between the intestinal lumen and the capillary blood and the structures between the sinusoidal space and the bile canaliculi in the liver, as well as the skin (see Fig. 4-1), are examples. Each of the interposing cellular membranes and spaces impede drug transport to varying degrees, and any one of them can be the slowest step, rate-controlling the overall transport process. In the skin, the stratum corneum is the major site of impedance. In the small intestine, it is the apical side of the epithelial cells, that is, the side facing the lumen. These complexities of structure make accurate extrapolation of the quantitative features of drug transport from one membrane to another difficult. Nonetheless, much can be gained by considering the general qualitative features governing drug transport across these “functional” membranes.
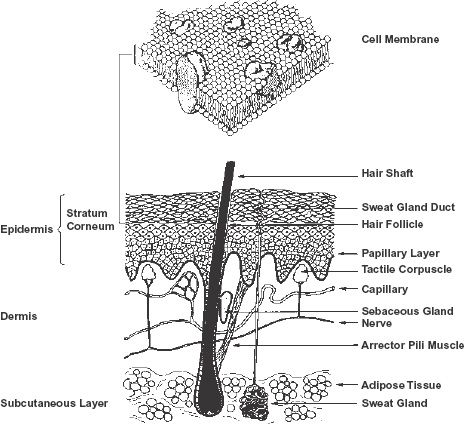
FIGURE 4-1. Functional membranes vary enormously in structure and thickness. They can be as thin as a single-cell membrane of approximately 1 × 10−6 cm thickness (top), to as thick as the multicellular barrier of the skin. This multicellular barrier extends from the stratum corneum to the upper part of the papillary layer of the dermis, adjacent to the capillaries of the microcirculation; a distance of approximately 2 × 10−2 cm (bottom). The cell membrane comprises a bimolecular leaflet, with a lipid interior and a polar exterior, dispersed through which are globular proteins, depicted as large solid irregular shaped bodies. (From: Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science 1972;175:720. Copyright 1982 by the AAAS; skin was kindly drawn by Mandy North.)
TRANSPORT PROCESSES
Drug transport can broadly be divided into transcellular and paracellular processes, as shown in Fig. 4-2. Transcellular movement, which involves the passage of drug through cells, is the most common route of drug transport. Some drugs, however, are too polar to pass across the lipoidal cell membrane and for them only the paracellular pathway, between the cells, is generally available. Other drugs move across some cell membranes by facilitative mechanisms.
PROTEIN BINDING
Before considering the determinants of the permeability of drug itself, a comment needs to be made about protein binding. Many drugs bind to plasma proteins and tissue components (discussed later in this chapter). Such binding is generally reversible and usually so rapid that equilibrium is established within milliseconds. In such cases, the associated (bound) and dissociated (unbound) forms of the drug can be assumed to be at equilibrium at all times and under virtually all circumstances. Only unbound drug is capable of diffusing through cell membranes. Proteins, and hence protein-bound drugs, are much too large to do so. Hence, the unbound concentration, not the total concentration, is the driving force for drug transport across a cell membrane.
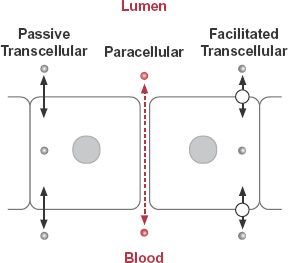
FIGURE 4-2. Movement of drugs across membranes occurs by paracellular and transcellular pathways. Paracellular movement (dashed colored arrow) of drug ( ) is influenced by the tightness of the intercellular junctions. The transcellular pathways can be divided into two categories: those in which the passage is by simple diffusion (solid arrows) and those in which facilitative mechanisms (arrows with a circle) are involved.
) is influenced by the tightness of the intercellular junctions. The transcellular pathways can be divided into two categories: those in which the passage is by simple diffusion (solid arrows) and those in which facilitative mechanisms (arrows with a circle) are involved.
DIFFUSION
One process by which drugs pass through membranes is diffusion, the natural tendency for molecules to move down a concentration gradient. Movement results from the kinetic energy of the molecules, and because no work is expended by the system, the process is known as passive diffusion.
To appreciate the properties of passive diffusion across a membrane, consider a simple system in which a membrane separates two well-stirred aqueous compartments. The driving force for drug transfer is the difference between the unbound concentrations in compartment 1, Cu 1, and compartment 2, Cu 2. The net rate of transport is:

The importance of the surface area of the membrane is readily apparent. For example, doubling the surface area doubles the probability of collision with the membrane and thereby increases the penetration rate twofold. Some drugs readily pass through a membrane, others do not. This difference in ease of penetration is quantitatively expressed in terms of the permeability, P. Note that the product P · SA has the units typical of flow, volume/time (cm3/min). With SA, surface area involved, having units of cm2, it is apparent that permeability has units of velocity, distance/time (cm/min).
Drug Properties Determining Permeability. Three major molecular properties affecting, and sometimes limiting, the passage of a drug across a given membrane are size, lipophilicity, and charge (or degree of ionization). These properties, together with the nature of the membrane and the medium on either side, determine the overall speed of movement of a compound across the membrane.
Molecular size has only a small impact on diffusion of substances in water. However, molecular size has a major impact on movement through membranes. This sensitivity to size is because of the relative rigidity of cell membranes, which sterically impedes drug movement. For some membranes, water-soluble materials cannot move through cells; instead, they move paracellularly through narrow channels between cells. Here, molecular size is the primary determinant of transport. To appreciate the impact of molecular size on paracellular transport, consider the passage of three water-soluble drugs, atenolol, oxytocin, and calcitonin-salmon across two membranes, the relatively loosely knit nasal membranes and the relatively tightly knit gastrointestinal membranes. Atenolol, a β -blocker used in the treatment of hypertension, is a small stable molecule (246 g/mol) that readily passes across the nasal membranes and is even reasonably well absorbed across the gastrointestinal membranes, with an oral bioavailability of 50%, facilitating its oral dosing. Oxytocin, a moderately sized cyclic nanopeptide (1007 g/mol) used to induce labor, also rapidly crosses nasal membranes paracellularly but is almost totally unable to cross the gastrointestinal membranes. Whereas calcitonin-salmon, a synthetic polypeptide of 32 amino acids (3432 g/mol) used in the treatment of postmenopausal osteoporosis, is so large that it is only 3% absorbed from a nasal spray and cannot pass across the intestinal epithelium. Furthermore, most nasally applied oxytocin and calcitonin are swallowed and undergo extensive degradation (digestion) by peptidases in the gastrointestinal tract, so that these drugs are not orally administered.
A second source of variation is lipophilicity. Generally, the more lipophilic the molecule the greater is its permeability but, as mentioned, size is also important. Small lipid-soluble, un-ionized drugs tend to traverse lipid membranes transcellularly with ease. This tendency and the effect of molecular size are shown in Fig. 4-3 for transdermal passage of various uncharged molecules. Notice, as mentioned above, as size increases permeability drops sharply. For example, for only a doubling of molecular weight (MW) from 400 to 800 g/mol for molecules with the same lipophilicity, permeability decreases by a factor of almost 2.5 log units, or 300-fold.
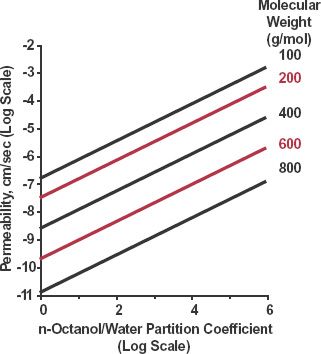
FIGURE 4-3. Permeability across skin as a function of molecular size and lipophilicity of neutral molecules. Lipophilicity is expressed as the n-octanol/water partition coefficient. Each line represents substances of the same molecular weight but different lipophilicity. Note, to offset the effect of reducing permeability on doubling of molecular weight, from 400 to 800 g/mol, lipophilicity has to increase by 4 log units, from 2 to 6, equivalent to a 10,000-fold increase in the partition coefficient. (From: Potts RO, Guy RH. Predicting skin permeability. Pharm Res 1992;9:633–669. Reproduced with permission of Plenum Publishing Corporation.)
The importance of lipophilicity and molecular size is also supported by observations of the movement of various drugs into the central nervous system (CNS), as shown in Fig. 4-4. The brain and spinal chord are protected from exposure to various substances. The protective mechanism was observed many years ago when various hydrophilic dyes, injected intravenously into animals, stained most tissues of the body, but not the CNS, which appeared to exclude them. Thus, the concept of high impedance to the movement of these substances into brain arose, namely, the blood-brain barrier. The barrier exists because of very tight junctions between the endothelial capillary cells as well as the presence of glial processes surrounding the capillaries highly resistant to polar substances. Figure 4-4 clearly shows that lipophilicity, as measured by the n-octanol/water partition coefficient, is a major determinant of the transport of drugs across the blood-brain barrier. The two major exceptions, the anticancer drugs vinblastine and vincristine, are moderately large molecules (molecular masses of 814 and 824 g/mol) and as stated above, size is also a major determinant of passage across membranes. The drugs are also substrates of efflux transporters.
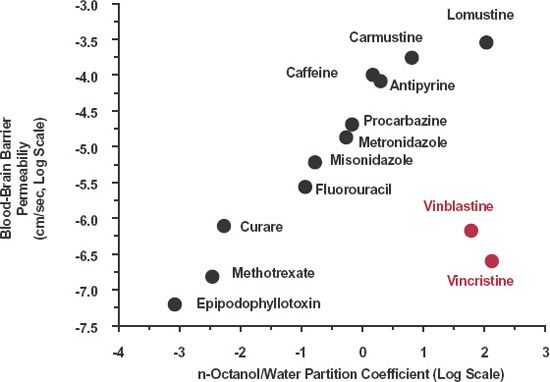
FIGURE 4-4. Relationship between permeability of a drug across the blood-brain barrier and its n-octanol/water partition coefficient. Generally, permeability increases progressively with increasing lipophilicity, but not always. For compounds such as vinblastine and vincristine (colored) the permeability is lower than expected, due in large part to their being substrates for the efflux transporter, P-glycoprotein. Both axes are logarithmic. (From: Greig N. Drug delivery to the brain by blood-brain barrier circumvention and drug modification. In: Neuwelt E, ed. Implication of the Blood-Brain Barrier and Its Manipulation. Vol. 1. Basic Science Aspects; 1989:311–367.)
Charge is the third major constraint to transmembrane passage. Again, there is considerable variation in the impedance of different membranes to charged molecules, but the effect of charge is, with a few exceptions (e.g., those involving paracellular transport across the blood capillary membranes and renal glomerulus), always large. The larger and more hydrophilic a molecule, the slower is its movement across membranes. Movement is slowed even more if the molecule is also charged. Some drugs are only partially charged (or ionized) at physiologic pH. Therefore, the degree of ionization is also important in determining movement across membranes.
Most drugs are weak acids or weak bases and exist in solution as an equilibrium between un-ionized and ionized forms. Increased total concentration of drug on the side of a membrane where pH favors greater ionization of drug has led to the pH partition hypothesis. According to this hypothesis, only un-ionized nonpolar drug penetrates the membrane, and at equilibrium, the concentrations of the un-ionized species are equal on both sides, but the total concentrations may be very different because of the differences in degree of ionization, as shown in Fig. 4-5. The topic of ionization and the pH partition hypothesis is expanded upon in Appendix B, Ionization and the pH Partition Hypothesis.
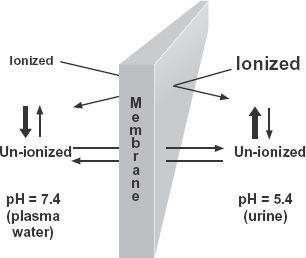
FIGURE 4-5. When a drug is a weak acid or weak base, its total concentration on one side of a lipophilic membrane may be very different from that on the other at equilibrium, if the pH values of the two aqueous phases are different. One mechanism producing this concentration difference is the pH partition hypothesis, which states that only the un-ionized form can cross the membrane and that the total concentration on each side at equilibrium depends on the degree of ionization. The side with greater ionization has the higher total concentration. For weak bases, the example shown, the total concentration is greater on the side with the lower pH; the opposite applies to weak acids. The relative concentrations of un-ionized and ionized drug on both sides of the membrane are shown by the font sizes. The weak base has a pKa of 6.4.
Most evidence supporting the pH partition hypothesis stems from studies of gastrointestinal absorption, renal excretion, and gastric secretion of drugs, all anatomical locations where pH is highly variable. The pH of gastric fluid varies between 1.5 and 7.0; that of intestinal fluids varies between 6.2 and 7.5, whereas urine pH varies between 5.0 and 7.5. Elsewhere in the body, changes in pH tend to be much smaller and to show less deviation from the pH of blood, 7.4. An exception is the acidic (pH 5) lysosomal region within cells where digestion of intracellular material takes place.
Despite its general appeal, the pH partition hypothesis fails to explain certain observations. Some small quaternary ammonium compounds (e.g., bethanechol chloride), which are always ionized, are absorbed and elicit systemic effects when given orally. Movement of these compounds through the gastrointestinal membranes occurs, although at a slow and unpredictable rate. Part of this movement is paracellular, but for many compounds, influx and efflux transporters are involved. For these reasons, quantitative prediction of the influence of pH on the movement of drugs across a membrane, based solely on the pH partition hypothesis, is often inaccurate.
Membrane Characteristics. Although molecular size, lipophilicity, and charge are generally key determinants of transmembrane passage of compounds, properties of the membrane are important as well. Some membranes, such as the renal glomerulus and blood capillaries of most tissues, are highly permeable to molecules up to 5000 g/mol in size with little effect of charge or lipophilicity. In these cases, drug transfer occurs paracellularly by movement through large fenestrations (windows) in the membrane. Movement of water through the membranes (a convective process) tends to augment the transport in the direction of flow. Table 4-1 lists membranes in general ascending order with regard to the influence of size (up to 5000 g/mol), lipophilicity, and charge on drug transport.
Another determinant of permeability is membrane thickness, the distance a molecule has to traverse from the site of interest (e.g., an absorption surface) to a blood capillary. The shorter the distance, the higher is the permeability. This distance can vary from about 0.005 to 0.01 μm (for cell membranes) to several millimeters (at some skin sites; see Fig. 4-1, lower part of figure).

Drug transport continues toward equilibrium, a condition in which the concentrations of the diffusing (generally unbound and uncharged) species are the same in the aqueous phases on both sides of the membrane. Movement of drug between these phases still continues at equilibrium, but the net flux is zero. Equilibrium is achieved more rapidly with highly permeable drugs, and when there is a large surface area of contact with the membrane, that is when the P · SA product is high (see Eq. 4-1).
Initially, when drug is placed on one side of the membrane, it follows from Eq. 4-1 that rate of drug transport is directly proportional to concentration (Fig. 4-6). For example, rate of transport is increased twofold when concentration of drug is doubled. Stated differently, each molecule diffuses independently of the other, and there is no upper limit to the rate of transport, unless the drug alters the nature of the membrane. Both absence of competition between molecules and lack of an upper limit to the rate of transport are characteristics of passive diffusion.
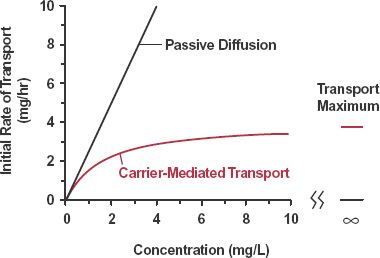
FIGURE 4-6. Initial rate of drug transport is plotted against the concentration of drug placed on one side of a membrane. With passive diffusion, the rate of transport increases linearly with concentration. With carrier-mediated transport, the rate of transport approaches a limiting value at high concentrations, the transport maximum.
CARRIER-MEDIATED TRANSPORT
Although many drugs are passively transported through cells, for many others the transport is facilitated, that is, the transport across the membrane is faster than expected from their physicochemical properties. Figure 4-7 shows several types of facilitated transport. The first example is passive facilitated diffusion in which movement is facilitated by a transporter, or transport system, which aids in speeding up the bidirectional process but does not change the condition at equilibrium. Such transporters are sometimes known as equilibrating transporters.
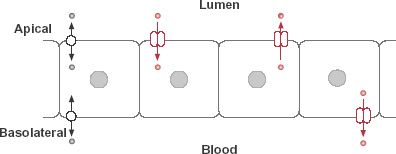
FIGURE 4-7. The intestinal epithelium, which exemplifies the general transport properties of membranes, forms a selective barrier against the entry of drugs into blood. Movement into (influx) and out of (efflux) the epithelial cells occurs by facilitative mechanisms, involving equilibrating transporters (bidirectional passive transport at either or both the apical and basolateral membranes) and concentrating transporters (in color). Concentrating transporters require energy and may involve influx, in which case the drug concentrates in the cell, or efflux, in which case drug is kept out of the cell.
Passive facilitated diffusion is exemplified by the gastrointestinal absorption and transmembrane passage into and out of tissue cells of many nucleosides and nucleobases. It is a passive process; the nucleosides and nucleobases move down a concentration gradient without expenditure of energy and, at equilibrium, the unbound concentrations across the membrane are equal. At high plasma drug concentrations, however, the rate of transport reaches a limiting value or transport maximum. This is a characteristic of facilitated transport processes, as shown in Fig. 4-6. Furthermore, in common with other carrier-mediated systems, passive facilitated transport is reasonably specific and is inhibited by other competing substrates of the same carrier. Drugs, so handled, include cytarabine, used to treat hairy cell leukemia, and gemcitabine, used in treating pancreatic cancer.
Additional types of facilitated transport, shown in Fig. 4-7 for intestinal transport, require energy and are capable of moving drug against an opposing concentration gradient. They are adenosine triphosphate (ATP) dependent and are examples of active transport systems, sometimes known as concentrating transporters. The direction of net movement may be either into the cell (influx transporter) or out of the cell (efflux transporter) and may occur on either the apical (lumen) or basolateral (blood) side of the membrane. Intracellular metabolizing enzymes may convert the drug to another substance before they both reach the blood. Apical efflux transporters and intracellular enzymes may, by concerted action, materially reduce systemic absorption, particularly for some drugs given orally.
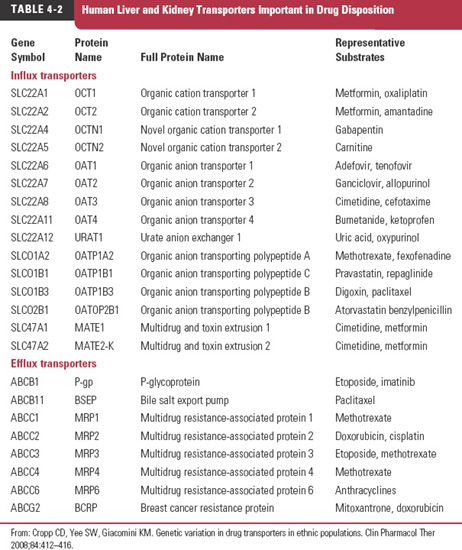
Efflux transporters also play a major role in removing metabolic end products and xenobiotic (foreign) substances from cells and organs. Our awareness of the therapeutic importance of efflux transporters was heightened when certain tumor cells were observed to be resistant to specific anticancer drugs. A transporter appeared to exclude many drugs from the cell; it was called the multiple drug-resistant receptor (MDR1). A specific glycoprotein, which resides in the cell membrane and is called permeability glycoprotein, or P-glycoprotein (170,000 g/mol), was found to be responsible. This ATP-dependent transporter is located in many organs and tissues. It plays a major role in the hepatic secretion into bile of many drugs, the renal secretion of many others, and the rate and extent of absorption of some drugs from the gastrointestinal tract. Many other drug transporters have since been identified. Examples of hepatic and renal transporters and their substrates are listed in Table 4-2. The location of these and other transporters and the processes in which they are involved in drug absorption and disposition are shown schematically in Fig. 4-8.
The CNS exemplifies, perhaps to the greatest extent, the consequences of the presence of carrier-mediated transport on drug distribution. The apparent lack of movement of many drugs across the blood-brain barrier is explained not only by the lipoidal nature of the barrier and the virtual absence of paracellular spaces, but also by the presence of efflux transporters. Many of these efflux transporters, P-glycoprotein being the most important identified so far, have the potential to keep the unbound concentration in the CNS relatively low compared to that in plasma, even at equilibrium.
To appreciate whether a transporter is likely to influence the relative concentration of drug on either side of a membrane, consider Fig. 4-9, which schematically shows three transport processes in brain. The dashed arrow is passive diffusion; the open arrow is passive facilitated diffusion, and the solid arrow is one involving an active efflux process. In Fig. 4-9A, the passive processes are fast and active efflux transport is relatively slow or absent. As a consequence, at equilibrium, the unbound concentration in brain is virtually the same as that in plasma. In Fig. 4-9B, the passive processes are slow compared to the efflux process so that the unbound concentration within the brain always remains low compared to that in blood. The apparent lack of entry into the CNS is shown in Fig. 4-10 for indinavir (Crixivan), a moderately large compound (MW = 712 g/mol) used for treating human immunodeficiency virus (HIV) infections.
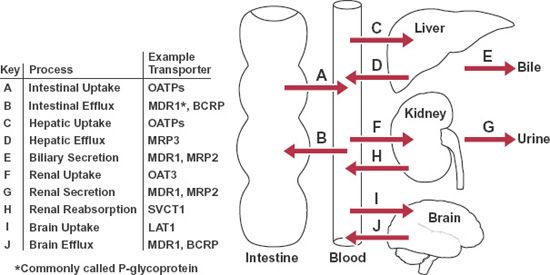
FIGURE 4-8. Selected transporters involved in intestinal absorption and in disposition of drugs within the liver, kidney, and brain. The names of these transporters and their general transport function within each of these tissues are identified.
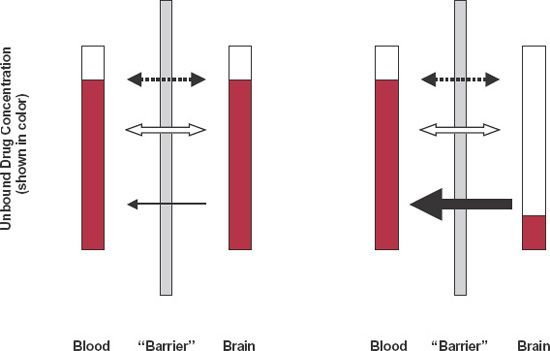
FIGURE 4-9. Substances enter the brain by simple diffusion (dashed arrow) and facilitated diffusion (open arrow). Even when active efflux transporters (solid arrow) are also present, the equilibrium ratio of unbound concentrations, brain to blood, can be close to one if the passive processes are inherently faster than the efflux transport (on left) or close to zero if the opposite is true (on right). For substances whose physicochemical properties (small in size, lipophilic, no charge) make them diffuse quickly, equilibrium tends to be rapidly achieved with left-hand condition prevailing.
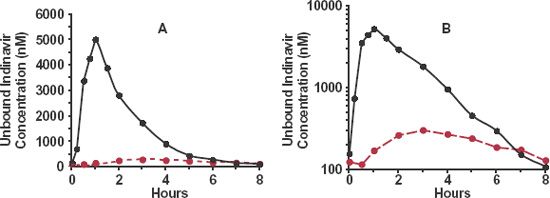
FIGURE 4-10. The mean steady-state unbound concentration–time profile of the HIV protease inhibitor indinavir in cerebrospinal fluid (colored) and plasma (black) over dosing interval during chronic oral administration of 800 mg every 8 hr in 8 symptom-free adults with HIV type 1 infection. A. Linear plot. B. Semilogarithmic plot. Note the much lower average unbound indinavir concentration in cerebrospinal fluid than in plasma. Also note that the unbound cerebrospinal fluid concentration peaks much later than does that of plasma and has a greatly reduced fluctuation. These observations are consequences of slow passive diffusion and efficient efflux, resulting in a slow and incomplete movement of drug into the cerebrospinal fluid. (From: Haas DW, Stone J, Clough LA, et al. Steady-state pharmacokinetics of indinavir in cerebrospinal fluid and plasma among adults with human immunodeficiency virus type 1 infection. Clin Pharmacol Ther 2000;68:367–374.)
Antihistamines further demonstrate the importance of drug transport and lipophilicity in drug action. Unlike the “first generation” antihistamines (e.g., chlorpheniramine, diphenhydramine, and hydroxyzine), which caused drowsiness, the “second generation” antihistamines (e.g., azelastine [Astelin], cetirizine [Zyrtec], fexofenadine [Allegra], and desloratadine [Clarinex]) are essentially devoid of sedative properties, not because they interact at a different receptor, or have a stimulating effect, but rather because they poorly penetrate the blood-brain barrier. This poor penetration is a consequence of their being good substrates for the efflux transporter, P-glycoprotein, as well as their being more hydrophilic than the “first generation” compounds.
REVERSIBLE NATURE OF TRANSPORT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


