Figure 1.1
Scheme of cardiac pain pathway. Pain originated from the heart is transmitted to a second neuron in the dorsal horn of the spinal cord, which carries out the signal to a third neuron in the thalamus, from where the signal is transmitted to cortical pain centres. The pain signal is modulated at various levels by intermediate neurons (gates) (Modified from Ref. [2])
Patients with predominant or exclusive silent myocardial ischemia, on the other hand, have some evidence of a defective processing of painful stimuli [6, 7]. Compared to patients with predominantly painful ischemia, they have been shown to have a higher threshold and tolerance for pain stimuli [6]. An increased release of endogenous opioids in the central nervous system has also been proposed as a possible mechanism, but assessment of plasma endorphins has given controversial results [8, 9].
Obstructive Coronary Atherosclerosis
Pathophysiology of Coronary Stenosis
The presence of stenosis caused by atherosclerotic plaques in epicardial conductive coronary vessels is the most frequent substrate for myocardial ischemia and angina.
In normal individuals, any increase in myocardial oxygen demand can only be met by a proportional increase of CBF. This is possible because coronary resistance in resting conditions is quite elevated, due to a high basal tone of small coronary arteries; the increase in myocardial oxygen demand induces dilatation of these vessels, thus reducing coronary resistance and increasing blood flow (metabolic CBF regulation). Of note, in normal conditions large epicardial conductive vessels do not offer an appreciable resistance (and therefore obstacle) to flow, which can increase up to five folds during elevated myocardial oxygen demand, as during maximal exercise.
When present, a significant coronary stenosis, instead, offers a resistance to blood flow able to cause a decrease of the driving pressure distal to the stenosis. As perfusion pressure is a major determinant of blood flow, the pressure drop distal to a stenosis tends to reduce blood flow in the supplied myocardial territory. In basal conditions this does not have any significant effect on myocardial perfusion because the pressure drop is compensated by a parallel reduction of distal coronary resistance, determined by dilatation of arteriolar vessels.1 This compensatory vasodilatation, however, also results in a reduction of coronary flow reserve (CFR) i.e., the magnitude of maximal increase of CBF, as compared to basal. The level of cardiac work at which it is not possible to further proportionally increase CBF to fully meet the increased metabolic requirements, thus resulting in myocardial ischemia (and angina), is defined ischemic (and angina) threshold.
Experimental studies [10] have shown that coronary stenosis usually starts to limit flow only when its lumen diameter is reduced at least by 50 %, while lower degrees of stenosis do not usually offer significant obstacle to blood flow at rest. Beyond this critical reduction of vessel lumen, however, any further increase of the stenosis causes a considerable reduction of the distal driving perfusion pressure, which, according to Poiseuille law,2 is inversely proportional to the fourth power of the minimum radius of the vessel lumen. Thus, increasing reductions in vessel diameter cause an exponential increase of trans-stenotic pressure gradient and a progressive reduction of maximal CBF, and therefore of CFR (Fig. 1.2). Of note, a reduction of 85–90 % of coronary artery diameter was required to cause myocardial ischemia at rest in dog. The relationship between the severity of coronary stenosis at angiography and the impairment of CFR has been confirmed in patients [11].
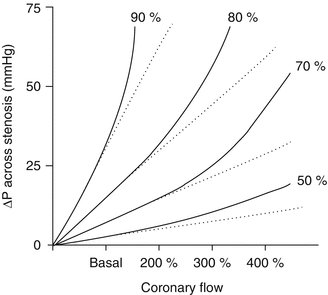
Figure 1.2
Relation between coronary blood flow and pressure gradient across a coronary stenosis. Post-stenotic pressure decreases progressively with the increase of stenosis severity and, for a given stenosis, it decreases markedly with increasing flow. The relation becomes curvilinear because of energy losses caused by blood flow turbulence across the stenosis (solid lines) (Modified from Ref. [10])
Myocardial ischemia consequent to an increase of myocardial oxygen demand, related to the presence of a coronary stenosis, typically involves subendocardial layers. The latter, indeed, present a lower flow reserve, and are therefore more vulnerable to ischemia compared to subepicardial layers. Subendocardial cells are indeed subject to the effect of diastolic intraventricular pressure, which increases their stretching and, therefore, according to Starling law, their systolic stress and contractility; as a result, oxygen consumption of subendocardial cells is slightly higher at rest, compared to that of subepicardial cells. Accordingly, blood flow in subendocardial layers is, in basal conditions, 15–20 % higher than that in subepicardial layers, which results in a lower flow reserve.
Moreover, subendocardial layers have greater susceptibility to ischemia also because subendocardial flow during diastole can be directly contrasted by the effects of intraventricular pressure and extravascular forces (Fig. 1.3) [10, 12], which exert scarce effect on subepicardial strata. Again, in normal heart, this disadvantageous condition does not have relevant effect on subendocardial flow, even in conditions of maximal oxygen demand, as during strenuous exercise. In presence of a flow-limiting coronary stenosis, instead, shortening of diastole (due to tachycardia) and a further increase of extravascular force (due to an increase of end diastolic left ventricular pressure that can result from ischemia), combined with the reduced driving pressure distal to the stenosis, can become crucial in favouring subendocardial ischemia during increased myocardial work and oxygen requirements.
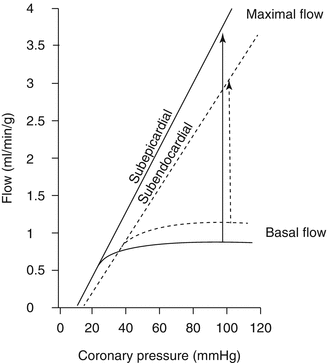
Figure 1.3
Basal flow, maximal flow and coronary flow reserve (CFR, arrows) in subendocardial (dotted lines) and subepicardial (continuous lines) layers of the left ventricle in relation to the driving pressure. CFR is lower in subendocardial layers, where resting blood flow is higher due to a higher basal oxygen consumption and maximal blood flow is lower due to extravascular resistance (Modified from Ref. [12])
In clinical practice the hemodynamic relevance of a coronary stenosis is assessed by coronary angiography, either visually or using quantitative measurements. However, the assessment of a coronary stenosis on the base of its angiographic severity only has several limitations: (1) coronary angiography is often inadequate to evaluate the tri-dimensional structure of a stenosis; (2) other factors may have relevance in determining the clinical effects of a stenosis, including the diameter of the original vessel, the length and the concentric or eccentric profile of the stenosis, as well as the presence of other stenoses in series; (3) the hemodynamic effects of the stenosis are scarcely correlated with its angiographic severity; the driving post-stenotic pressure, indeed, also depends on other factors, including basal arterial pressure, collateral vessel development, dynamic modulation of the stenosis and of distal microcirculation, and extravascular resistance.
A method now considered more reliable to assess the hemodynamic significance of a coronary stenosis is the measure of fractional flow reserve (FFR) [13]. FFR is calculated as the ratio between the mean pressures distal and proximal to the stenosis during maximal vasodilatation, usually induced by intracoronary administration of adenosine. An FFR <0.80 is believed to identify a flow-limiting stenosis, able to cause myocardial ischemia during increased myocardial oxygen consumption. However, the results of this method can also be influenced by several factors, including collateral circulation, basal microvascular state, as well as preload and afterload at the time of assessment, and clinical trials failed to demonstrate a significant usefulness of FFR measurement for improving major clinical events by coronary revacularization [14]. Accordingly, at present, the consequences of a stenosis in the clinical setting seem to be more adequately evaluated by assessing the presence, degree and extension of myocardial ischemia in the correspondent myocardial territory by appropriate imaging stress tests.
Finally, the hemodynamic significance of a stenosis can also be assessed by obtaining a measure of CFR, expressed as the ratio between CBF velocity during maximal coronary vasodilatation induced by a vasodilator drug (usually adenosine) and CBF velocity at baseline, as assessed by intracoronary Doppler recording. CFR >2.5 as measured by this method is usually believed to be normal [15]. Measurements of CBF velocity, however, are also subject to variations and significantly depend on the state of coronary microcirculation and basal CBF as well.
Factors Influencing Epicardial Stenosis
Dynamic Changes
In only a minor number of cases coronary stenoses are fixed, i.e., do not show any variation at their site of vessel lumen able to significantly influence blood flow. These stenoses are usually associated with a quite stable and predictable pattern of myocardial ischemia and angina. Nevertheless, coronary stenoses often present at their site vasomotor changes that are able to significantly modify their severity, and therefore CFR and angina threshold, thus resulting in a variable and much less predictable angina pattern. The tendency to dynamic changes of a stenosis can be assessed by evaluating the vasomotor response to intracoronary administrations of vasodilator or vasoconstrictive substances [16].
Collateral Circulation
When a stenosis, or even a chronic coronary occlusion, causes a reduction of blood flow, myocardial perfusion can still be guaranteed by the development of coronary collateral vessels, which connect a normal vascular territory to that supplied by the obstructed vessel. Collateral blood flow, indeed, increases post-stenotic pressure in the supplied territory, and therefore improves CBF and CFR.
Collateral vessels mainly develop from pre-existing intercoronary arterial anastomoses, which are stimulated to grow by the pressure gradient that develops between their origin from the normally supplied vessels and their termination at the site of vessels supplied by the stenotic vessel [17]. These preexisting anastomoses progressively transform into mature small vessels, with a final diameter of 20–200 μm, through a complex process that involves their initial widening and remodelling, subsequent proliferation of endothelial cells and smooth muscle cells, with development of a smooth muscle coat. Collateral vessels may also develop as new vessels, but this mechanism does not seem to contribute significantly to collateral circulation in man. Collateral circulation can be assessed by coronary angiography and shows significant variations among patients, the reasons for which remain poorly known (Fig. 1.4) [18].
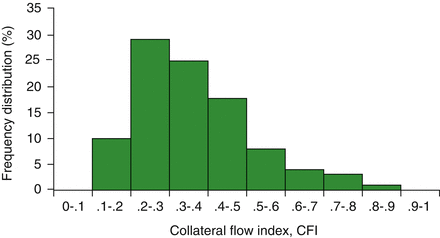
Figure 1.4
Distribution of collateral flow index in patients with total occlusion of a coronary artery vessel; a large variability of collateral blood flow is observed (Modified from Ref. [18])
As specified above, collateral blood circulation may contribute significantly to the variability of ischemic and angina threshold in individual patients; blood flow in collateral vessels may indeed undergo significant variations in different conditions, due to influences by neural, humoral and autacoid stimuli. Furthermore, collateral circulation may have protective effects in several conditions; it can be associated with scarce or no ischemia during coronary occlusion caused by thrombosis or spasm, a minor extension of myocardial infarct following persistent thrombotic occlusion, and a better functional recovery after coronary by-pass surgery in patients with left ventricular dysfunction [19].
Coronary Steal
Coronary steal is a particular mechanism of myocardial ischemia and angina occurring when blood flow to a myocardial region supplied by a severely stenotic vessel becomes insufficient because of a diversion of a significant quote of blood flow towards another myocardial region, following maximal arteriolar dilation occurring in the latter. Coronary steal typically occurs in subendocardial layers as a result of microvascular dilatation in the subepicardial circulation supplied by the same stenotic vessel. The reduction of coronary resistance in the subepicardium, indeed, favours blood flow towards these areas, which, together with the consequent reduction of the driving perfusion pressure, will compromise blood flow to the subendocardium, which, as discussed above, present a reduced vasodilator reserve (trasmural or vertical steal) (Fig. 1.5) [20].
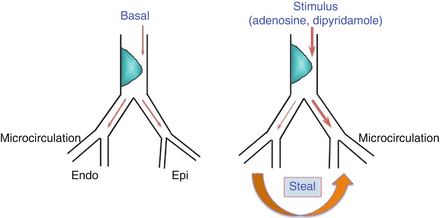
Figure 1.5
Myocardial ischemia caused by transmural coronary flow steal. In presence of a stenosis, subendocardial flow (Endo) at baseline is guaranteed by a higher degree of microvascular dilatation, as compared to subepicardial layers (Epi). During vasodilator stimuli, subepicardial vessels are maximally dilated causing a steal of blood from subendocardial vessels
Coronary steal can also occur in myocardial areas which depend on collateral circulation to maintain a valid blood flow; maximal dilatation in the territory of the supplying normal (or even mildly stenotic) coronary vessel will indeed result in a parallel reduction of blood flow in the collateral vessels, thus causing myocardial ischemia in the supplied territory (intercoronary or lateral steal) [21].
Typically, coronary steal can be induced by arteriolar dilatory drugs (e.g., adenosine, dipyridamole), an effect utilized in pharmacological stress tests for diagnosis of coronary artery disease [22]. Some other drugs, as xanthines, on the other side, can contrast the induction of coronary steal. Xanthines prevent arteriolar dilatation in the territory responsible for the steal by antagonizing the effects of adenosine (the main mediator of arteriolar dilatation during myocardial ischemia) and by inducing α-mediated vasoconstriction through inhibition of norepinephrine re-uptake by sympathetic nerve endings [23].
Coronary Artery Spasm and Vasoconstriction
The term coronary artery spasm (CAS) specifically indicates a sudden, intense, occlusive or subocclusive constriction of an epicardial coronary artery, which determines a dramatic primary reduction of CBF, usually resulting in transmural myocardial ischemia. CAS should be taken distinct from abnormally increased vasoconstriction, which can be defined as an enhanced, but not occlusive/subocclusive, response, as compared to a normal response, to low-medium doses of constrictor stimuli.
Recurrent CAS typically results in the clinical syndrome of Prinzmetal’s variant angina [24], which is characterized by angina attacks occurring at rest and associated with ST segment elevation on the electrocardiogram (ECG) (Fig. 1.6). The vasospastic mechanism of variant angina was clearly demonstrated by coronary angiographic studies performed during spontaneous angina attacks [25]. CAS, however, can also be a major or contributing mechanism, in some cases, of a clinical presentation of acute coronary syndrome. Enhanced vasoconstriction, on the other side, may contribute to cause subendocardial ischemia at rest or favour exercise-induced angina when severe or occurring at the site of sub-critical stenoses.
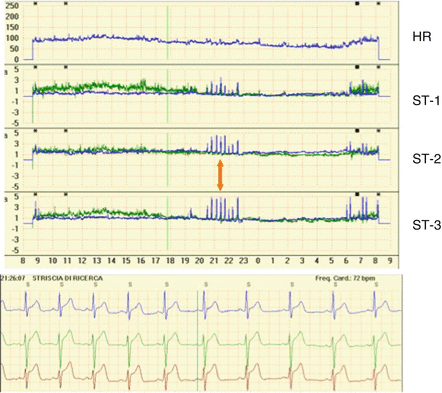
Figure 1.6
Twenty-four-hour Holter ECG recording in a patient with variant angina. Top panel: The four graphs represent the 24-h trends of heart rate (HR) and ST-segment (ST) level (blue lines) and slope (green lines) of 3 ECG leads (CM5-CM3-modified aVF), respectively. Several short episodes of ST-segment elevation can be seen between 20:00 and 23:00 and between 6:00 and the end of the recording. Bottom panel: ECG strip taken at the time indicated by the arrow, with the patient reporting chest pain in the diary; typical ST-segment elevation can be observed in all three ECG leads
CAS can occur at the site of a stenosis (either minor or severe) or in angiographically normal coronary arteries. Furthermore, CAS can be localized to a segment of an epicardial artery (focal spasm) (Fig. 1.7), involve two or more segments of the same artery (multifocal spasm) or different epicardial arteries (multivessel spasm), or may also involve diffusely one or multiple coronary branches (diffuse spasm) [26].
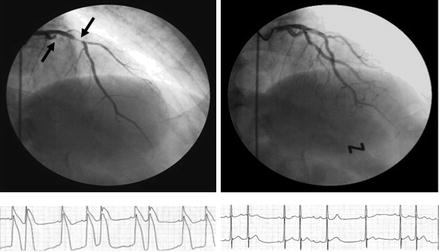
Figure 1.7
Occlusive spasm of the left circumflex coronary artery and subocclusive spasm of the left anterior descending coronary artery (arrows) during coronary angiography (left, top), associated with dramatic ST segment elevation at monitoring ECG leads (left, bottom). Complete resolution of spasm (right, top) and ST segment elevation (right, bottom) after intravenous nitrate administration (Modified from Ref. [28])
CAS usually lasts a few minutes, but in some cases it can be prolonged and cause myocardial injury. Furthermore, CAS can cause local mechanical damage of the endothelium and vessel wall, thus favouring coronary thrombosis and acute myocardial infarction [26]. Importantly, transmural ischemia caused by CAS can be complicated by malignant ventricular arrhythmias [27].
Although its causes and mechanisms are still poorly known, CAS results, anyway, from the interaction of two components (Fig. 1.8): (1) an abnormality of a coronary artery which makes it hyper-reactive to vasoconstrictor stimuli, and (2) a vasoconstrictor stimulus able to trigger the spasm of the hyper-reactive coronary segment [28].
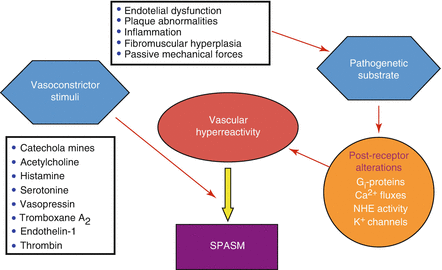
Figure 1.8
Pathophysiologic mechanisms of coronary artery spasm. See text for details (Modified from Ref. [28])
Several factors suggest that CAS substrate requires a hyper-reactivity of vascular smooth muscle cells (SMCs): (1) vasoconstrictor stimuli that induce CAS in the involved coronary segments are unable to induce spasm in other coronary segments of the same patients as well as in patients with other forms of angina [29, 30]; (2) CAS can usually be elicited by various stimuli that act through different receptors and cellular pathways [31], which also suggests a post-receptorial, intracellular alteration of SMC hyper-reactivity; (3) coronary SMC hyper-reactivity to constrictor stimuli, resulting in spasm, can be induced in experimental models [32].
Experimental and clinical data suggest that an increased activity of Rho-kinase, an intracellular enzyme which increases myosin light chains sensitization to intracellular Ca++, can be involved in the SMC hyperreactivity responsible for CAS [33, 34]. Rho-kinase was indeed found increased in patients with vasospastic angina [34] and its inhibitor fasudil prevented acetylcholine-induced CAS in these patients [35]. Other cellular pathways, however, have been suggested to be potentially involved in CAS, including protein kinase C activation, lack of SUR-2, a component of the ATP-dependent K+ channel, and increased membrane Na+-H+ exchanger [28].
Some studies have suggested that a significant endothelial dysfunction might also contribute to CAS, as the consequent impairment of vasodilation favours the response to vasoconstrictor stimuli [36]. Of note, several drugs able to cause spasm through direct stimulation of vascular smooth muscle cells (i.e., acetylcholine, serotonin, histamine) in normal vessels cause endothelium-mediated vasodilation. Thus, in presence of endothelial dysfunction, their release in the vessel wall can lead to vasoconstriction or CAS. Furthermore, endothelial activation might also facilitate CAS due to endothelial release of vasoconstrictor substances, mainly endothelin-1 [37].
Other mechanisms have been proposed for susceptibility to CAS of coronary arteries, including passive mechanical collapse in presence of an eccentric stenosis, medial fibromuscular hyperplasia, adventitial release of vasoconstrictor substances from neural termination or inflammatory cells [38] and magnesium deficiency, but their role is likely limited to specific cases; in some patients an excessive consumption of alcohol or use of some drugs (e.g., cocaine, amphetamines, marijuana, 5-fluorouracile, capecitabine, sumatripan, etc.…) can result in susceptibility to CAS [28]. Among traditional cardiovascular risk factors, instead, only smoking has been found to show a significant association with CAS occurrence [39].
Finally, other factors shown to be potentially able to cause spasmogenic vascular changes include acute inflammation [38, 40], increased oxidative stress [41] and genetic factors, which have mainly concerned genes encoding for NO synthase [42] or adrenergic receptors [43].
As previously observed, a large number of stimuli can usually trigger spasm when acting at the site of a hyper-reactive coronary artery, although they are in most cases not promptly identifiable. They include a raise of either sympathetic or parasympathetic tone. In clinical practice sympathetic outflow seems responsible for CAS when the latter is induced by exercise or cold pressor test [44, 45], as well as substances (e.g., cocaine, amphetamines) known to increase sympathetic drive and/or sensitization to catecholamines of vascular smooth muscle cells [46].
Acetylcholine, the neurotransmitter of parasympathetic fibres, on the other side, is known to induce CAS in patients with vasospastic angina through stimulation of muscarinic receptors on smooth muscle cells [47]. In patients with vasospastic angina, attacks often occur during the night, suggesting a role for vagal activity [26]; ischemic episodes occurring at night, however, might be related to a transient surge of sympathetic, rather than parasympatheitic, activation [48].
Vasoconstrictor substances, able to trigger spasm, can be released by activated platelets, endothelium or inflammatory cells. Finally, hyperventilation, by inducing alkalosis, is a well-known stimulus for CAS and can be a possible under-recognized trigger of spasm in at least some patients with vasospastic angina [49].
Coronary Microvascular Dysfunction
Experimental studies clearly showed that myocardial ischemia can be induced by abnormalities in the coronary microcirculation. The intracoronary infusion of the vasoconstrictor endothelin-1 or fLMP in dog and rabbit, respectively, was indeed able to induce myocardial ischemia in the absence of any changes in large coronary vessels, thus suggesting constriction of small coronary arteries [50, 51].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


