CHAPTER 12 Macrocytic anemia
Macrocytic anemias fall into two categories: those associated with megaloblastic hemopoiesis and those associated with normoblastic hemopoiesis.1–3 The most common causes of megaloblastic hemopoiesis are vitamin B12 or folate deficiency. Disruption of vitamin B12 or folate metabolic pathways may also cause this type of disturbed hemopoiesis as may vitamin B12 or folate-independent mechanisms that interfere with DNA synthesis.
Megaloblastic hemopoiesis
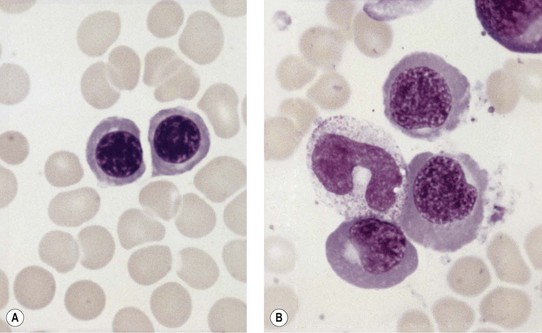
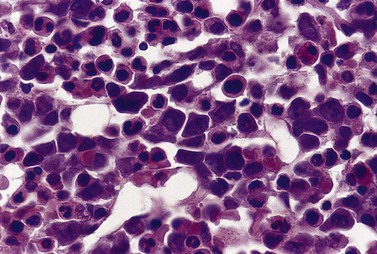
Paul Ehrlich first used the term megaloblast in 1880 to describe a morphologically abnormal erythroblast seen in the bone marrow of patients with untreated pernicious anemia. It was subsequently found that megaloblasts occur in many other conditions (see Boxes 12.1 and 12.2). Megaloblastic erythropoiesis is characterized by three features: 1) erythroblasts that are larger than normal at all stages of maturation; 2) a dissociation between cytoplasmic and nuclear maturation leading to early and late polychromatic erythroblasts with well-hemoglobinized (i.e. polychromatic) cytoplasm having nuclei containing considerably less mature and more open appearing chromatin than their normal counterparts (Figs 12.1 and 12.2); and 3) the generation of larger than normal macrocytes typically oval in shape (Fig. 12.3). In megaloblastic erythropoiesis there is also an increased relative number of early and late polychromatic erythroblasts with dysplastic features (see Chapter 5), an increased number of basophilic erythropoietic cells relative to more mature erythroblasts (Fig. 12.4) and erythroid hyperplasia, giving the overall appearance of a maturation arrest of the bone marrow. The severity of each of these morphologic abnormalities increases with increasing severity of the anemia.
Box 12.1 Vitamin B12-related and folate-related causes of macrocytosis with megaloblastic erythropoiesis
Vitamin B12-related
Box 12.2 Vitamin-B12-independent and folate-independent causes of macrocytosis with megaloblastic erythropoiesis
Abnormalities of nucleic acid synthesis
Megaloblastic erythropoiesis is substantially ineffective and the extent of ineffectiveness is generally proportional to the extent of the anemia. The ineffectiveness of megaloblastic erythropoiesis results from an abnormality of the red cell precursors during the S-phase of the cell cycle, which leads to apoptosis with consequent phagocytosis by bone marrow macrophages of a substantial proportion of the early and late polychromatic megaloblasts.4 The biochemical lesion is believed to occur during the S-phase of the cell cycle, uniformly throughout the maturational sequence among precursors undergoing mitosis. Hence, vitamin B12-deficient or folate-deficient early polychromatic megaloblasts appear morphologically to become arrested at all stages of the cell cycle.
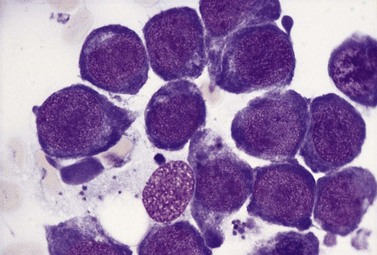
Usually patients with megaloblastic erythropoiesis also show morphologic abnormalities (‘megaloblastic changes’) in cells of the granulocyte series. The two most striking abnormalities are the formation of giant metamyelocytes in the marrow (Fig. 12.5) and the presence of hypersegmented neutrophil granulocytes in the blood (Fig. 12.6). The giant metamyelocytes are 17–30 µm or more in diameter and usually have long horseshoe-shaped nuclei, sometimes with one or more bud-like protuberances. In addition, these cells may contain cytoplasmic vacuoles, nuclear perforations or unevenly stained chromatin. Giant metamyelocytes have DNA contents in the entire range between the diploid (2c) and tetraploid (4c) values. This seems to result from abnormal development in promyelocytes and myelocytes that have been arrested or retarded during their progress through the cell cycle, in much the same way as occurs in the erythroid series. Most of these defective giant cells may be phagocytosed by bone marrow macrophages but a few undergo nuclear segmentation and develop into giant polymorphonuclear leukocytes known as ‘macropolycytes’ (Fig. 12.7); these have hyperdiploid DNA contents. By contrast, hypersegmented neutrophils have diploid DNA contents and are presumed to be derived from relatively normal-looking metamyelocytes with diploid DNA contents. In either case, the underlying problem appears to be a qualitative defect in DNA synthesis.
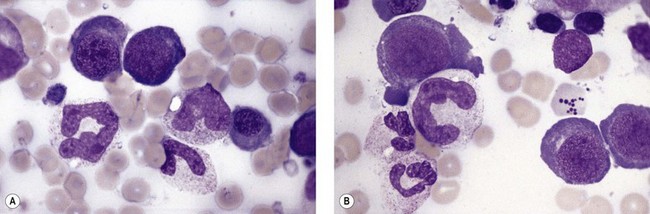
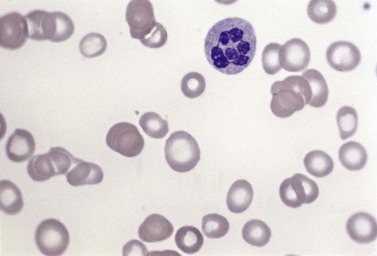
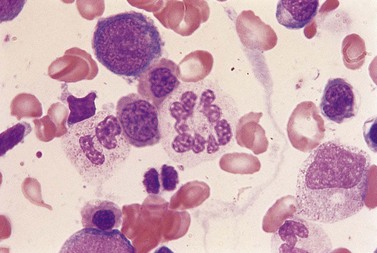
Fig. 12.7 Hypersegmented macropolycyte and a normal-sized neutrophil with two nuclear segments from the bone marrow smear of a patient with pernicious anemia. The nuclear and cytoplasmic areas of the macropolycyte are similar to those of the giant metamyelocytes in Fig. 12.5. May–Grünwald–Giemsa stain. × 1000.
Vitamin B12-related and folate-related causes of megaloblastic anemia
The vitamin B12– and folate-related causes of macrocytosis with megaloblastic erythropoiesis are given in Box 12.1.
Vitamin B12
The vitamin B12 molecule consists of two parts aligned at right angles to each other: 1) a planar corrin nucleus (containing four pyrrole rings); and 2) the ribonucleotide of 5,6-dimethylbenzimidazole. A cobalt atom is located at the center of the corrin nucleus and is coordinately bonded to the four pyrrole rings and below the pyrrole plane, to one of the nitrogen atoms of the ribonucleotide as well as to an upper ligand above the pyrrole plane such as methyl, deoxyadenosyl, cyano or hydroxo. The two naturally occurring B12 coenzymes contain the methyl or deoxyadenosyl group and are known as methylcobalamin and deoxyadenosylcobalamin, respectively. In nature, vitamin B12 is synthesized exclusively by prokaryotic microorganisms. Synthesis by bacteria resident in the gut serves as the main source of B12 in ruminant herbivores and in others through deliberate or incidental coprophagia. Other animals and man obtain B12 by consuming foods of animal origin, including dairy products. Vegetables and fruits are devoid of vitamin B12 except through contamination by bacteria. A mixed diet contains about 5–30 µg vitamin B12 per day and 1–3 µg of this are absorbed. The vitamin in food is largely protein-bound and is released from its bound state within the stomach by the action of the proteolytic enzyme pepsin. Most of the released B12 rapidly attaches to a B12-binding protein found in saliva and gastric juice known as R-binder, a haptocorrin-like binder. Subsequently, B12 is released from the R-binder in the jejunum as a result of the alkaline pH and degradation by pancreatic trypsin. The released B12 then combines with intrinsic factor, a glycoprotein secreted by the parietal cells of the fundus and body of the stomach (Fig. 12.8). The vitamin B12-intrinsic factor complex, which is resistant to digestion, passes down to the distal portion of the ileum where absorption takes place via a specific receptor, termed cubam, on the brush border of the mucosal cells. This receptor is coded for by two genes, cubilin and amnionless.5 There is a mucosal delay of a few hours before the absorbed vitamin B12 enters the portal blood. Most of the newly absorbed vitamin B12 in portal blood and 20–30% of total circulating B12 is attached to a specific B12 binding protein known as transcobalamin (TC) (previously transcobalamin II) although most of the vitamin B12 in the blood is bound to haptocorrin (previously transcobalamin I).
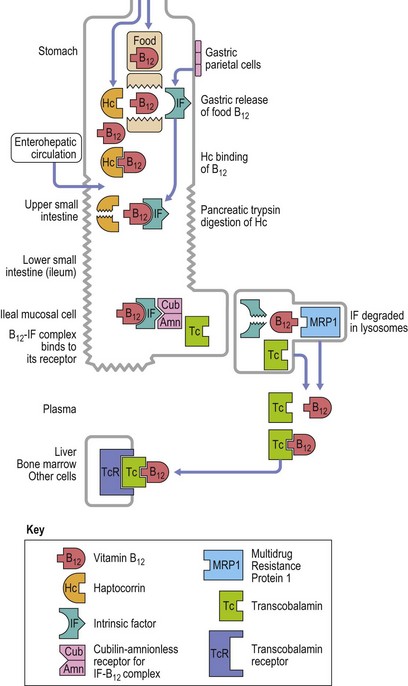
Fig. 12.8 Diagram showing vitamin B12 absorption, cellular uptake and enterohepatic recirculation.
(Adapted from Hoffband AV, Green R in Hoffband AV, Catovsky D, Tuddenham EGD, Eds. Postgraduate Hematology 5th Edition, Oxford, Blackwell Ltd).
Vitamin B12 is found mainly in the liver, the hepatic stores being 2–5 mg. The absorption of 1–3 µg vitamin B12 per day balances an inevitable daily loss of the same magnitude. Loss occurs largely in the urine and feces via desquamation of epithelial cells and in the bile. There is an enterohepatic circulation of vitamin B12:6 about 3–6 µg is excreted daily into the intestinal tract, mainly in the bile, of which all but about 1 µg is reabsorbed in the terminal ileum. If intrinsic factor is absent or ileal absorption is defective, there is failure to conserve vitamin B12 secreted in the bile.7
The biochemical mechanisms by which vitamin B12 deficiency leads to its main clinical consequences of anemia, peripheral neuropathy and subacute combined degeneration of the spinal cord remain uncertain.2 Only two reactions are known to require vitamin B12 in man. These are: 1) the isomerization of methylmalonyl coenzyme A to succinyl coenzyme A, which occurs in the mitochondria and is dependent on deoxyadenosylcobalamin; and 2) the cytosolic methylation of homocysteine to methionine, which requires the enzyme homocysteine-methionine methyl transferase (methionine synthase), the methyl donor 5-methyltetrahydrofolate (methyl-THF) and the coenzyme methylcobalamin. During the latter reaction the methyl-THF is converted to THF and impairment of this reaction in bone marrow cells results in defective methylation of deoxyuridylate to thymidylate due to a decreased availability of 5,10-methylenetetrahydrofolate (5,10-methylene-THF) (Fig. 12.9). Defective thymidylate synthesis is thought to lead to defective DNA synthesis, caused by misincorporation of uracil into DNA and, consequently, to the development of megaloblastic hematopoiesis and anemia.2 The mechanism by which impairment of the transferase reaction results in decreased levels of 5,10-methylene-THF is still controversial. Some have suggested that this impairment results in ‘trapping’ of intracellular folates in the form of 5-methyl-THF which cannot be converted to 5,10-methylene-THF.8,9 Others have considered that the important consequence of the impairment of the transferase reaction is the failure of methionine synthesis which in turn results in a reduced availability of formate and inadequate formylation of tetrahydrofolate, formyltetrahydrofolate being the necessary substrate for polyglutamate synthesis, required for 5,10-metheylene-THF-polyglutamate formation (Fig. 12.9).10
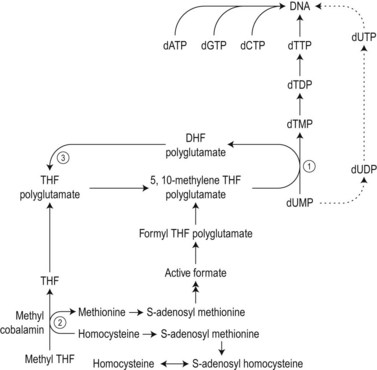
The biochemical mechanism underlying the neurologic damage induced by vitamin B12 deficiency is unclear.2,11,12 The balance of evidence suggests that the neurologic damage may result from a failure to methylate basic proteins in myelin sheaths secondary to a failure of the synthesis of S-adenosylmethionine from methionine as a consequence of the decreased methylcobalamin-dependent conversion of homocysteine to methionine (Fig. 12.9).5 The clinicopathologic features of subacute combined degeneration of the cord and the peripheral neuropathy caused by vitamin B12 deficiency are described in the section on pernicious anemia. These neurologic abnormalities have also developed in patients with vitamin B12 deficiency due to veganism, partial or total gastrectomy, abnormal overgrowth of small intestinal bacterial flora, ileal resection and the Imerslund–Gräsbeck syndrome. A severe peripheral neuropathy has also been reported in inadequately-treated patients with congenital transcobalamin II deficiency.
Causes of vitamin B12 deficiency (Box 12.1)
Veganism.13,14 As vitamin B12 is not found in vegetables or fruit, strict vegetarians (vegans – i.e. those who do not eat meat or fish and little or no milk, milk products or eggs) have a very low to apparently absent dietary intake of this vitamin. The largest group of vegans is found amongst the Hindus. Although the majority of vegans have low serum vitamin B12 levels, most vegans have normal hematological values, including mean cell volumes (MCV), and appear to be in good health. It appears that the enterohepatic circulation of vitamin B12 enables avid conservation of the very small quantities of vitamin B12 absorbed from the diet and likely derived through microbial contamination of food to ensure adequate supplies of the vitamin to marrow and other cells despite the extremely low vitamin B12 stores. However, some vegans with a low serum B12 level develop a megaloblastic anemia which responds to treatment with either oral or parenteral vitamin B12 and a few suffer from vitamin B12 neuropathy. Breast-fed infants of vegan mothers may develop vitamin B12 deficiency during the first year of life.15 People on a predominantly vegetarian diet who consume some dairy products may also develop low serum B12 levels and it is possible that some of them have an additional acquired defect in the ability to release B12 from food (see below).
Pernicious anemia.3 In this condition impaired vitamin B12 absorption and vitamin B12 deficiency result from a marked reduction in the secretion of intrinsic factor secondary to gastric atrophy caused by autoimmune destruction of gastric parietal cells. The vitamin B12 deficiency may lead to anemia, neurologic damage or both. Though the disease is common in people of Northern European origin, it also occurs in Africans, Asians, Chinese and other races. The diagnosis may be missed or delayed in these ethnic groups because of masking of the hematological features of macrocytosis by coexistent thalassemia or iron deficiency. Pernicious anemia is uncommon before the age of 30 years and the incidence increases with advancing age, most affected individuals being 50–70 years old. According to the older literature, the overall prevalence in the UK is about 1 per 1000 of the general population, but rises to 1% after the age of 60 years. A study among the multiethnic population of Los Angeles, California, aged 60 years or over showed that the prevalence of mild cobalamin deficiency due to undiagnosed pernicious anemia was at least 2.7% in women and 1.4% in men; the prevalence was 4.3% in black women and 4.0% in white women.16 The male:female ratio in pernicious anemia is about 1 : 1.5. A family history of pernicious anemia is present in about 20% of patients. Autoimmune diseases (thyroid diseases, vitiligo, hypoparathyroidism and hypofunction of the adrenal glands) are more common in patients with pernicious anemia and their relatives than in the general population, and patients show a slightly higher than normal incidence of the blood group A. Pernicious anemia patients have an increased incidence of adenocarcinoma of the non-cardia portion of the stomach, and surprisingly, also have an increased risk of esophageal squamous cell carcinoma.17 There is a more substantial increase in gastric carcinoid tumors but these tend to be of the benign type.18
The above data indicate that there is a genetic predisposition to the development of gastric atrophy and that autoimmune mechanisms are involved. However, the absence of intrinsic factor and parietal cell antibodies in some cases of pernicious anemia suggests that these antibodies may be a consequence rather than the cause of the damage to the gastric mucosa. Studies of a murine model of autoimmune chronic atrophic gastritis suggest that cell-mediated rather than humoral immunity may be involved.19 Indeed, the gastric mucosa in pernicious anemia shows infiltration with plasma cells and lymphocytes, with an excess of CD4+ cells. These histological changes revert to a more normal appearance and both acid and intrinsic factor (IF) secretion increase following steroid administration.
When pernicious anemia was first recognized over a century ago, the disease was diagnosed at an advanced stage, usually with severe megaloblastic anemia, other cytopenias and progressive neurologic abnormalities. Today, with the availability of automated full blood counts (including MCV), automated serum B12 and folate and red cell folate assays as well as assays for metabolites that rise in B12 deficiency, particularly methylmalonic acid, B12 deficiency is usually diagnosed at an early stage. In one study, 44% of B12-responsive patients had hematocrit values within the normal range and 36% had MCVs within the normal range.20 In developing countries patients still present with severe megaloblastic anemia.
Symptoms and signs. Symptoms are of slow onset and may include tiredness, weakness, dyspnea, a sore tongue and gastrointestinal disturbances (anorexia, nausea, vomiting, dyspepsia, constipation, diarrhea) and loss of weight. There may be slight jaundice, a low-grade pyrexia and slight enlargement of the spleen. Neurologic symptoms, which affect only some patients, usually begin in the lower limbs and are symmetrical. The most frequent of these are paresthesiae in the extremities, difficulty in walking and muscle weakness. Others include poor vision, stiffness of the limbs, impotence and in advanced cases, impairment of bladder and rectal control. Neurologic signs include sensory loss (particularly loss of position and vibration senses) which is worse in the legs, ataxia, positive Romberg sign, impairment of memory and, less commonly, the features of spastic paraplegia. The severity of neurologic impairment correlates inversely with the hematocrit;21 about a quarter of cases with B12 neuropathy are not anemic and a slightly lower proportion do not have a high MCV.22
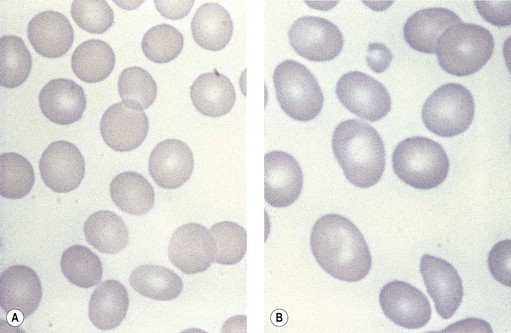
Laboratory investigations and treatment. Patients diagnosed at the earliest stages have a hemoglobin level and MCV within the reference range. Those diagnosed slightly later first present with an increased red cell distribution width (RDW) followed by a high MCV (macrocytosis) and high mean cell hemoglobin (MCH) and still with a hemoglobin level within the reference range. Later, there is both macrocytosis and mild to severe anemia, depending on the duration and severity of the deficiency. In anemic patients, the reduction of the red cell count is more marked than that of the hemoglobin level. Usually some of the macrocytes are oval in shape (see Fig. 12.3). With increasing anemia, the red cells also show an increasing degree of anisocytosis and poikilocytosis. The poikilocytes include tear-drop-shaped and irregularly-shaped cells as well as small red cell fragments (schistocytes). There is a rough inverse correlation between the degree of anemia and the MCV; unusually mild degrees of macrocytosis for the extent of anemia are found when there are many small red cell fragments (usually in patients with Hb less than 7 g/dl) and in patients with coexistent iron deficiency, chronic disorder or thalassemia syndrome.23 The circulating neutrophil granulocytes of most cases show a tendency towards hypersegmentation of their nuclei (Fig. 12.6), more than 3% of the neutrophils containing five or more nuclear segments, although hypersegmentation may not be present in early stages of B12 deficiency.24 There may be mild neutropenia or thrombocytopenia, particularly in severely anemic patients.
Serum bilirubin level of the unconjugated type may be slightly elevated and the serum lactate dehydrogenase is usually increased, often dramatically. The serum iron is high but falls within 48 h of a single injection of vitamin B12. The serum vitamin B12 level is below the normal reference range in 95–97% of cases. However, a low serum vitamin B12 level should be considered as presumptive rather than definitive evidence of vitamin B12 deficiency as low levels are also seen in about one-third of patients with folate deficiency; this is especially the case in countries not practising folic acid fortification. Furthermore, low vitamin B12 levels may be found in normal individuals, particularly in pregnant women or in the elderly, sometimes without any hematologic, neurologic or even biochemical disturbances attributable to a deficiency of this vitamin. Red cell folate levels are low in the majority of patients with pernicious anemia and normal in the remainder. The serum folate level is low in 10% of cases presumably because of secondary intestinal malabsorption of folate resulting from intestinal megaloblastosis caused by B12 deficiency and may be high in 20–30%, resulting from the methyl-folate trap, discussed above (Fig. 12.9). Parietal cell antibodies, directed against the α and β subunits of the proton pump (H+, K+ ATPase) of the gastric parietal cell,19 are found in the serum in about 85% of patients and IgG anti-intrinsic factor antibodies in 55%. The gastric juice contains an IgA antibody against intrinsic factor in about 60% of cases.
Because of the impairment of the mitochondrial adenosylcobalamin-dependent conversion of methylmalonyl CoA to succinyl CoA, plasma methylmalonic acid (MMA) levels are increased in vitamin B12 deficiency (but not in folate deficiency). In addition, the impairment of the methylation of homocysteine, which is dependent both on methylcobalamin and methyl-THF, leads to an increase in plasma homocysteine (HCYS) levels in either vitamin B12 or folate deficiency. Nearly all clinically confirmed cases of vitamin B12 deficiency have increased levels of plasma MMA or HCYS or both.25 Whereas increased MMA levels are highly specific for vitamin B12 deficiency, increased HCYS levels are less specific, being found in patients with impaired renal function, hypothyroidism, B6 deficiency, alcoholism and some inborn errors of homocysteine metabolism.26
Vitamin B12 absorption tests, such as the Schilling test (now unavailable), showed impaired absorption of an orally-administered physiologic dose of radiolabeled vitamin B12; the impaired absorption was improved by the simultaneous oral administration of intrinsic factor. Newer methods, replacing the Schilling test, are now under development. The definitive diagnosis of pernicious anemia requires the presence of circulating antibodies to intrinsic factor. Intrinsic factor antibodies, though insensitive, are highly specific, being virtually confined to pernicious anemia. Demonstration of impairment of intrinsic factor production was achieved indirectly by performing a Schilling test, with and without intrinsic factor.3,5,27 There is gastric achlorhydria with raised levels of serum gastrin. Although the diagnosis of pernicious anemia cannot be sustained in the absence of achlorhydria, gastric aspiration is rarely performed nowadays. Therefore the presence of achlorhydria is established indirectly by demonstrating elevated levels of gastrin or pepsinogen in the blood. However, severe achlorhydria caused by simple atrophic gastritis is not uncommon in elderly subjects with adequate intrinsic factor secretion. Parietal cell antibodies are also not specific for pernicious anemia, being found in a small percentage of healthy individuals (2% of those less than 30 years of age and 16% of those greater than 60 years) and in a higher proportion of individuals with various disorders such as myxedema, Graves’ disease, iron deficiency anemia and gastritis without pernicious anemia. Because of the present difficulty of arriving at a definitive diagnosis of pernicious anemia, the diagnosis must often be presumptive, after exclusion of other causes of megaloblastic hemopoesis or neurological abnormalities. Frequently, management of such patients is empirical and because vitamin B12 treatment is inexpensive and safe, it is instituted without a definitive diagnosis. Ideally, if there is no objective improvement in the patients condition, then B12 treatment should be withdrawn. If efficacious, then it is important to maintain treatment, even if symptoms have not returned.
Patients with pernicious anemia may be initially treated with six intramuscular injections, each of 1 mg hydroxocobalamin, over 2–12 weeks to replenish body stores of vitamin B12. This should be followed by 1 mg hydroxocobalamin intramuscularly every 3 months, throughout life. In the USA, cyanocobalamin is the form of B12 used. As it is not retained as well as hydroxocobalamin, it should be given monthly for maintenance. High dose daily oral B12 may be used as an alternative since there is passive absorption of B12.28,29 Though inefficient (1% of the dose given is absorbed), doses of 1–2 mg daily suffice to maintain adequate vitmain B12 levels. Hematologic abnormalities are rapidly and completely reversed by vitamin B12 therapy. Neurologic symptoms of recent onset may improve considerably over 6–12 months.
Total or partial gastrectomy.30,31 Total gastrectomy results in the removal of the intrinsic factor-secreting cells and therefore inevitably leads to vitamin B12 depletion and eventually to megaloblastic anemia due to vitamin B12 deficiency. This anemia appears 2–10 years after the operation depending on the size of the B12 store and the extent of gastric resection. These factors determine the time taken for normal vitamin B12 stores to be depleted after the abrupt cessation or drastic diminution of vitamin B12 absorption. It is best to commence regular B12 therapy soon after the gastrectomy. The probability of developing vitamin B12 deficiency is a function of the amount of stomach resected. In most cases, the vitamin B12 deficiency appears to result from a combination of the loss of intrinsic factor-secreting mucosa at operation and the subsequent atrophy of the remainder of the gastric mucosa. The deficiency may also result from a failure to release vitamin B12 from food due to a deficiency of acid and gastric pepsin (see below). In a few cases, and particularly when blind loops have been created (as happens following a Polya type gastrectomy), the vitamin B12 deficiency may be the consequence of the development of an abnormal intestinal bacterial flora. The frequency with which gastrectomy is now carried out has markedly reduced since the advent of successful medical treatments for peptic ulceration and Helicobacter pylori infection. However, the widespread use of H blockers and proton pump inhibitors to reduce acid secretion has contributed to food B12 malabsorption (see below). In addition, the obesity epidemic in Western countries has led to a growth in the practice of gastric reduction surgery which may be complicated by nutrient, including vitamin B12 malabsorption.32 This malabsorption is caused by both a failure to digest and release food B12 and a diminution in intrinsic factor production. The clinical picture in such patients is further complicated by multiple nutrient deficiencies including that of copper, which can produce a myeloneuropathy resembling that seen in vitamin B12 deficient myelopathy as well as anemia, which may be macrocytic.33
Congenital intrinsic factor deficiency or mutation.34–36 Patients with this rare group of disorders present with megaloblastic anemia in childhood and usually during the first 3 years of life. The syndromes are characterized by the total absence of intrinsic factor or the production of a mutant intrinsic factor molecule which binds but fails to promote vitamin B12 absorption, However, in contrast to pernicious anemia, hydrochloric acid and pepsin are present in gastric juice, there is a normal-looking gastric mucosa, and parietal cell and intrinsic factor antibodies are absent. Inheritance is autosomal recessive and heterozygotes appear clinically normal.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


