I. NORMAL ANATOMY. Lymph nodes are the most widely distributed collections of lymphoid tissue within the lymphoreticular system, which also includes the thymus, tonsils, adenoids, spleen, and Peyer patches. Due to their easy accessibility, lymph nodes are the most frequently examined lymphoid tissue for a lymphoreticular disorder. Microscopically, the lymph node shows four compartments: The most obvious are the primary and secondary follicles, which are usually found near the capsule, surrounding the follicles and extending deeper into the node is the paracortex. The third and fourth compartments represent the medullary region and the sinuses, respectively (e-Fig. 43.1).*
II. GROSS EXAMINATION, TISSUE SAMPLING, AND HISTOLOGIC SLIDE PREPARATION. Fresh lymphoid tissue should be examined by gross inspection, touch preparation, or frozen section examination to assess whether the:
A. Tissue represents adequate sampling, and
B. Tissue needs to be allocated for various ancillary studies essential to a correct diagnosis. The fresh lymph node should be cut perpendicularly along the long axis, and material for ancillary studies procured as follows:
1. Wet touch preparations fixed in 95% alcohol or formalin, for H&E or Papanicolaou staining
2. Air-dried touch preparations for Giemsa or Wright-Giemsa staining, cytochemistry (myeloperoxidase, nonspecific esterase, etc.), or cytogenetics [i.e., fluorescence in situ hybridization (FISH)]
3. Rapidly frozen tissue for immunohistochemistry, cytochemistry, or genetic analysis
4. Fresh tissue (in RPMI 1640 medium or saline) for flow cytometry
5. Sterile fresh tissue for microbial cultures or cytogenetic analysis (karyotyping or FISH)
6. Thin-shaved tissue rapidly fixed in glutaraldehyde for electron microscopy
7. Paraffin-embedded tissue after fixation for routine H&E-staining, immunohistochemistry, and special stains [e.g., Giemsa, periodic acid-Schiff (PAS), elastin, trichrome, Leder, etc.]
Procuring tissue for histology takes priority over other studies. The most commonly used fixative for permanent sections is 10% neutral buffered formalin. B5 fixative is commonly used in addition to 10% neutral buffered
formalin because of the sharp nuclear detail it produces. However, this mercuric chloride-based fixative is very expensive and poses an environmental hazard. Furthermore, molecular studies cannot be carried out on B5-fixed paraffin-embedded tissue, because it will generally yield poor polymerase chain reaction (PCR) amplification results.
III. DIAGNOSTIC FEATURES OF COMMON BENIGN DISEASES OF LYMPH NODES. In reactive lymphadenopathy, there are five different architectural patterns to be recognized, with many showing a mixed pattern of response.
A. Follicular hyperplasia (e-Fig. 43.2) is characterized by an increase in number and size of B-cell germinal centers and is common in lymph node-draining sites of chronic inflammation. This pattern is also present in syphilitic lymphadenitis where, in addition to the marked lymphoid hyperplasia, thickening of the capsule by chronic inflammation, fibrosis, and neovascularization with arteritis and phlebitis, and a marked plasma cell infiltrate in the medullary region predominate. Rheumatoid lymphadenopathy and acute human immunodeficiency virus (HIV) lymphadenitis are other examples of marked follicular hyperplasia.
B. Diffuse (paracortical) hyperplasia shows expansion of the T-cell paracortical areas. This pattern is commonly seen in viral lymphadenitis [Epstein-Barr virus (EBV), cytomegalovirus (CMV), herpes] and vaccinia lymphadenitis revealing an expansion of the paracortex with increased immunoblasts, imparting a mottled appearance. Follicular hyperplasia and sinus dilation are often concurrent findings in this entity, resulting in a mixed pattern of lymphoid hyperplasia. Phenytoin lymphadenopathy represents a relatively pure diffuse hyperplasia showing an expanded paracortical T-zone with numerous large immunoblasts, eosinophils, plasma cells, and neutrophils.
C. Sinus hyperplasia describes increased cellularity within the medullary sinuses of lymph nodes. Sinus histiocytosis is seen in numerous nonspecific responses to chronic inflammation, as well as in lymph nodes draining solid tumors. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman) disease is characterized by markedly dilated sinuses filled with CD68+, S100+ histiocytes showing emperipolesis. Lipophagic reactions causing sinus histiocytosis with accumulation of phagocytosed fat include mineral oil ingestion, Whipple disease, and lymphangiography procedures. Prominent vacuoles in the histiocytes can generate a signet-ring cell histiocytosis, a pattern that should not be confused with signet-ring-cell carcinoma. Finally, vascular transformation of lymph node sinuses (e-Fig. 43.3) shows a sinus pattern, and it is important to distinguish this entity from Kaposi sarcoma (e-Fig. 43.4). The latter can be distinguished from the former by the proliferation of spindle-shaped Kaposi sarcoma cells forming cleft-like vascular spaces that contain erythrocytes, many of which are extravasated.
D. Granulomatous lymphadenopathy describes formation of epithelioid granulomas in lymph nodes. Caseating granulomas are epithelioid granulomas that form central necrosis and caseation and are typically seen in Mycobacterium tuberculosis lymphadenitis. Special stains for mycobacteria in paraffin-embedded tissue (Ziehl-Neelson, Fite-Faraco) will detect the bacilli as bright red, slender, beaded microorganisms (e-Figs. 43.5 and 43.6). Mycobacterium leprae lymphadenitis and histoplasma lymphadenitis are other examples of caseating granulomatous inflammation. Necrotizing, noncaseating granulomas are present in cat-scratch disease (e-Fig. 43.7) caused by Bartonella henselae, in which suppurative granulomas with stellate microabscesses surrounded by palisading histiocytes predominate. Kikuchi-Fujimoto lymphadenopathy is characterized by necrotizing granulomas containing karyorrhectic debris, but lacking neutrophils; this form of necrotizing lymphadenitis characteristically occurs in young Asian women. Nonnecrotizing, noncaseating granulomas are characteristic of sarcoidosis lymphadenopathy (e-Fig. 43.8), composed primarily of epithelioid histiocytes with scattered multinucleated giant cells, lymphocytes,
and plasma cells. This type of epithelioid granuloma can also be seen in draining lymph nodes of Crohn’s disease.
E. Acute lymphadenitis is an acute inflammation in lymph nodes draining an infected focus. Acute lymphadenitis is almost exclusively bacterial in nature, and morphologic features range from focal infiltration by neutrophils to necrosis and suppuration with abscess formation.
It should be mentioned that in most cases of benign reactive lymphadenopathy, a combination of more than one architectural pattern is present in the same lymph node.
IV. INCIDENCE AND EPIDEMIOLOGY OF NON-HODGKIN LYMPHOMAS
A. B-cell lymphomas. B-cell lymphomas constitute the vast majority of lymphomas in North America and Europe, accounting for nearly 90% of all lymphomas. Diffuse large B-cell lymphoma (DLBCL) (∽31%) (e-Fig. 43.9) and follicular lymphoma (∽22%) (e-Fig. 43.10) are the most common types. Immunosuppression, specifically due to HIV infection and immunosuppressive therapy to prevent graft versus host disease, is associated with a markedly increased incidence of mature B-cell lymphomas, particularly DLBCL and Burkitt lymphoma (e-Fig. 43.11).
Some of the low-grade B-cell lymphoproliferative disorders include follicular lymphoma grades 1 and 2, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) (e-Fig. 43.12), nodal and extranodal marginal zone Bcell lymphoma (e-Fig. 43.13), and lymphoplasmacytic lymphoma, which are generally indolent, but incurable and usually present in a disseminated stage with bone marrow involvement. Mantle cell lymphoma (e-Figs. 43.14 and
43.15) and DLBCL represent “intermediate-grade” B-cell lymphomas that generally show a more aggressive clinical behavior, but are potentially curable. The same applies to high-grade B-cell lymphomas, which include Burkitt lymphoma and precursor B-lymphoblastic leukemia/lymphoma.
B. T-cell lymphomas. Mature T-cell and natural killer (NK)-cell malignancies are rare, accounting for only 10% to 12% of all non-Hodgkin lymphoma (NHL), and usually are more aggressive than B-cell lymphomas. The most common subtypes are peripheral T-cell lymphoma, unspecified (˜4% of all adult NHLs and ˜30% of peripheral T-cell lymphomas) (e-Fig. 43.16) and anaplastic large cell lymphoma (˜3% of all adult NHLs) (e-Fig. 43.17). In general, T-cell and NK-cell malignancies are much more common in Asia and are linked to viral infection with EBV (NK-cell lymphomas) (e-Fig. 43.18) and human T-cell leukemia virus (HTLV-1) (adult T-cell leukemia/lymphoma) (e-Fig. 43.19). The current 2008 World Health Organization classification of lymphoid malignancies is summarized in Table 43.1.
V. PATHOPHYSIOLOGY OF NON-HODGKIN LYMPHOMAS
A. B-cell lymphomas. Mature B-cell malignancies mimic stages of normal B-cell differentiation; therefore, classification is generally based on their morphologic and immunophenotypic resemblance to the normal B-cell counterpart (Fig. 43.1) (Best Pract Res Clin Haematol. 2005;18:11). Normal B-cell development begins in the bone marrow where precursor B-lymphoblasts undergo immunoglobulin VDJ gene rearrangement and develop into IgM+, IgD+ naïve B-cells with surface immunoglobulin light chain and CD5 expression. These resting B-cells circulate in the blood and occupy primary follicles and mantle zones of secondary follicles. Malignant counterparts of CD5+ naïve Bcells are believed to be CLL and mantle cell lymphoma. Upon antigen stimulation, naïve B-cells undergo blastic transformation, migrate into the center of the primary follicle, and form the germinal center. These centroblasts are large lymphoid cells with vesicular chromatin and several eccentrically located nucleoli. They express BCL-6 and CD10 but switch off BCL-2 protein expression, therefore becoming susceptible to death through apoptosis. Centroblasts undergo intense proliferation that is accompanied by somatic hypermutation of their rearranged variable-region genes, giving rise to cells carrying receptors with high affinity for the stimulating antigen. This leads to a large pool of B-lymphoid cells with intraclonal diversity. Centroblasts then mature into centrocytes, representing intermediate-sized lymphoid cells with irregular, cleaved nuclei and inconspicuous nucleoli. Centrocytes with mutations resulting in decreased affinity for antigen die by apoptosis, whereas centrocytes with high affinity for antigen are rescued from apoptosis by reexpressing BCL-2. Malignant counterparts of germinal center-derived B-cells are follicular lymphoma, a subset of DLBCL, and Burkitt lymphoma. Via interactions with follicular dendritic cells through CD23 and with T lymphocytes through CD40 ligand (CD40L), centrocytes switch off BCL-6 expression and differentiate into either memory cells or plasma cells. Memory cells are found in the marginal zone of follicles; they typically express immunoglobulin M (IgM) and lack expression of IgD, CD5, CD10, or CD23. Plasma cells home to the bone marrow; they lack surface immunoglobulin and pan-B-cell expression, but express CD79a, CD138, and cytoplasmic IgG or IgA. Neither memory cells nor plasma cells undergo further somatic hypermutations. Marginal zone B-cell lymphoma corresponds to postgerminal center cells, possibly memory cells of marginal zone type. Plasma cell myeloma is the malignant counterpart of bone marrow-homing IgG- or IgA-producing plasma cells.
TABLE 43.1 2008 WHO Classification of Lymphoid Neoplasms
Mature B-cell neoplasms
Chronic lymphocytic leukemia/small lymphocytic lymphoma
B-cell prolymphocytic leukemia
Splenic B-cell marginal zone lymphoma
Hairy cell leukemia
Splenic B-cell lymphoma/leukemia, unclassifiable
Splenic diffuse red pulp small B-cell lymphoma
Hairy cell leukemia-variant
Lymphoplasmacytic lymphoma
Heavy chain diseases (gamma heavy chain/mu heavy chain/alpha heavy chain diseases)
Plasma cell neoplasms
MGUS
Plasma cell myeloma
Solitary PC of bone
Extraosseous PC
Monoclonal immunoglobulin deposition diseases
Extranodal marginal zone B-cell lymphoma of MALT lymphoma
Nodal marginal zone B-cell lymphoma
Follicular lymphoma
Primary cutaneous follicle center lymphoma
Mantle cell lymphoma
DLBCL, NOS
T-cell/histiocyte-rich large B-cell lymphoma
Primary DLBCL of the CNS
Primary cutaneous DLBCL, leg type
EBV positive DLBCL of the elderly
DLBCL associated with chronic inflammation
Lymphomatoid granulomatosis
Primary mediastinal (thymic) large B-cell lymphoma
Intravascular large B-cell lymphoma
ALK positive large B-cell lymphoma
Plasmablastic lymphoma
Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease
Primary effusion lymphoma
Burkitt lymphoma
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL
Mature T-cell and NK-cell neoplasms
T-cell prolymphocytic leukemia
T-cell large granular lymphocytic leukemia
Aggressive NK-cell leukemia
EBV positive T-cell lymphoproliferative diseases of childhood
Systemic EBV+ T-cell lymphoproliferative disease of childhood
Hydroa vacciniforme-like lymphoma
Adult T-cell leukemia/lymphoma
Extranodal NK-/T-cell lymphoma, nasal type
Enteropathy-associated T-cell lymphoma
Hepatosplenic T-cell lymphoma
Subcutaneous panniculitis-like T-cell lymphoma
Mycosis fungoides
Sézary syndrome
Primary cutaneous CD30+ T-cell lymphoproliferative disorders
Primary cutaneous peripheral T-cell lymphomas, rare subtypes
Primary cutaneous gamma-delta T-cell lymphoma
Primary cutaneous CD8 positive aggressive epidermotropic cytotoxic T-cell lymphoma
Primary cutaneous CD4 positive small/medium T-cell lymphoma
Peripheral T-cell lymphoma, NOS
Angioimmunoblastic T-cell lymphoma
Anaplastic large cell lymphoma, ALK positive
Anaplastic large cell lymphoma, ALK negative
From: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. Used with permission.
B. T lymphocytes. Two major classes of T lymphocytes exist on the basis of the structure of the T-cell receptor (TCR). Approximately 95% of all T-cells are αβ T-lymphocytes that can be subdivided into CD4+ helper T-cells or CD8+ cytotoxic T-cells. Only 5% of T-cells are γ δ cells, which are primarily found in the splenic pulp and intestinal epithelium and are not MHC restricted in their function, because they do not express CD4 or CD8. The TCR is composed of either the αβ or γ δ chains, each consisting of an external variable (V) and constant (C) domain. The TCR is complexed with CD3 that contains γ, δ, and ε chains. NK cells do not have a complete TCR, but usually express the ε chain of the CD3 in the cytoplasm; therefore, NK cells will stain positively with a polyclonal antibody to CD3. The malignant counterparts of γ δ T-cells are believed to be hepatosplenic T-cell lymphoma (e-Fig. 43.20) and enteropathytype T-cell lymphoma (e-Fig. 43.21). NK cells share some immunophenotypic markers and functions with cytotoxic CD8+ T-cells; these include expression of CD2, CD7, CD8, CD56, CD57, granzyme B, perforin, and T-cell intracellular antigen (TIA-1).
Due to their broad cytologic spectrum, absence of immunophenotypic markers of monoclonality, and general lack of specific genetic abnormalities, clinical presentation plays a major role in the subclassification of T-cell and
NK-cell malignancies (Fig. 43.2). For example, hypercalcemia is associated with adult T-cell leukemia; hemophagocytic syndrome occurs more frequently in cytotoxic T-cell or NK-cell malignancies; persistent severe neutropenia is a relatively common clinical feature in T-cell granular lymphocyte leukemia (e-Fig. 43.22); and systemic symptoms of edema, pleural effusion, ascites, arthritis combined with anemia, and polyclonal hypergammaglobulinemia are relatively specific for angioimmunoblastic T-cell lymphoma (e-Fig. 43.23).
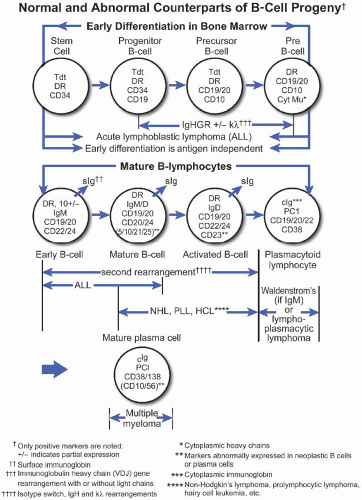
Figure 43.1 Brief overview of normal B-cell progeny and malignant counterparts.
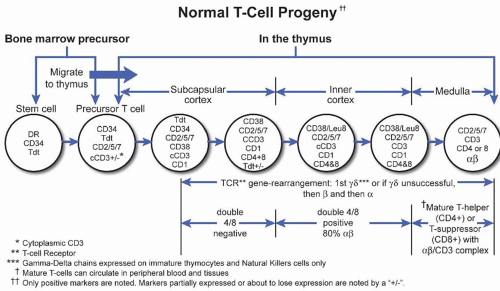
Figure 43.2 Brief overview of normal T-cell progeny.
TABLE 43.2 An Algorithm for the Workup of Lymphomas
Cell type: Are the cells lymphoid?
In a tissue section, what is the low-power architecture?
Cell size
Are the majority of cells small (same size as a lymphocyte)? Or large (∽3 times as big as a lymphocyte, or even bigger)?
Is there an even mixture of small and large cells? Are they anaplastic?
Nuclear shape
Do the cells have round nuclei with a smooth contour (noncleaved)? Or a round shape with a bumpy or notched contour (irregular)? Or a variable shape with deeply folded or grooved nuclei (cleaved)?
Histologic grade
Is necrosis present? Is there a “starry sky” appearance? Are mitoses easy to find?
Lineage (by flow cytometry or immunohistochemistry)
Are the cells of B lineage (generally CD20+) or T lineage (generally CD3+)?
Maturation
Are the cells at a precursor (TdT+) or a mature (TdT−) stage of maturation?
If mature and of B lineage, are they prefollicular, follicular, postfollicular, or effector cell stage?
If mature and of T lineage, are they at a thymic or postthymic/effector cell stage?
Disease specific markers
Do the cells express cyclin D1, bcl6, ALK-1, and so on?
VI. A RATIONAL APPROACH TO GENERAL LYMPHOMA WORKUP. The classification of lymphomas may appear difficult; however, a systematic approach can be used to efficiently narrow the differential diagnosis for most cases. Needless to say, knowledge of clinical history and a basic complete blood count are required to guide the initial steps of the evaluation (e.g., to direct the immunophenotypic workup of the sample by flow cytometry), because tissue sections are often not available until the next day. The general algorithm (see Table 43.2) starts with the low-power appearance of the tissue section. Is the architecture nodular, follicular, diffuse, or mixed? What is the cell size; small (close to a normal lymphocyte), medium, or large (about three times the size of a normal lymphocyte)? What is the nuclear shape (cleaved nuclei of follicular lymphoma or round nuclear contours of chronic lymphocytic lymphoma)? Is the process high grade (necrosis, starry sky appearance, high mitotic rate)? The workup continues with a determination of the lineage and stage of maturation. As mentioned above, flow cytometry is usually extremely helpful in delineating the lineage, provided care is taken to save fresh tissue in RPMI medium and order appropriate markers; alternatively, immunohistochemistry can be performed on formalin-fixed, paraffin-embedded tissue. The workup concludes with evaluation of disease-specific markers such as cyclin D1 and anaplastic large cell lymphoma kinase (ALK-1), which can only be evaluated by immunohistochemistry in tissue sections.
Antigen markers useful in delineating and subclassifying lymphoid malignancies are the following:
A. Leukocyte marker. CD45 [also known as leukocyte common antigen (LCA)] is expressed on all leukocytes.
B. Markers of immaturity
1. TdT (terminal deoxynucleotidyl transferase, a specialized DNA polymerase; nuclear expression present only in pre-B and pre-T lymphoblasts)
2. CD34 (present on pluripotent hematopoietic stem cells and progenitor cells of many lineages)
3. CD10 (CALLA, common acute lymphoblastic leukemia antigen); expressed on marrow pre-B-cells and mature follicular center B-cells)
4. CD22 (present on pre-B-cells)
5. cµ (cytoplasmic µ heavy chain)
C. Primarily B-cell associated markers
1. CD19 (present on marrow pre-B-cells, mature B-cells, but not on plasma cells; no paraffin-reactive antibody available)
2. CD20 (present on marrow pre-B-cells, mature B-cells, but not on normal plasma cells)
3. CD79a (present on mature and pre-B-cells, as well as on plasma cells)
4. CD22 [transmembrane molecule, present on mature B-cells (cCD22) and pre-B-cells (cCD22)]
D. Markers helpful in subclassifying mature B-cell lymphomas
1. CD5 (expressed on all T-cells and small subset of B-cells, expressed on neoplastic CLL and mantle cell lymphoma cells)
2. CD10
3. CD11c (expressed in high levels on monocytes, macrophages, and NK cells, as well in moderate levels on granulocytes; expressed on subsets of T and B-cells)
4. CD23 (present on activated mature B-cells)
5. CD38 (primarily expressed on mature B-cells and plasma cells)
6. CD43 (leukosialin, expressed on the surface of all leukocytes except resting B-cells; CD43 expression in B-cell lymphomas is highly correlated with CD5 and is therefore an indicator of aberrant B-cell populations)
7. BCL-6 (expressed normally in germinal center lymphocytes in the normal lymph node; it is distributed in a pattern reciprocal to that of BCL-2)
8. BCL-2 (present on T-cells and normal mantle B lymphocytes; aberrantly expressed in majority of B-cell lymphomas)
9. Cyclin D1 (cell cycle regulatory protein rearranged through the t(11;14) of mantle cell lymphoma; also expressed weakly in hairy cell leukemia and plasma cell dyscrasias; nuclear protein detectable in paraffin-embedded sections)
10. CD138 (Syndecan-1, expressed on most cases of myeloma, but also present in carcinomas)
E. Markers of clonality: κ and λ immunoglobulin light chains
F. Primarily T cell- and NK cell-associated markers:
1. CD1 (expressed on cortical thymocytes and Langerhans cell histiocytes)
2. CD2 [present on all T-cells (thymic and peripheral T-cells) and NK cells]
3. CD3 (lineage-specific marker for T-cells; cytoplasmic form also expressed in NK cells)
4. CD5 (expressed on all T-cells and a small subset of B-cells)
5. CD7 (expressed on all T-cells and a small subset of myeloid precursor cells)
6. CD8 (present on cytotoxic subset of peripheral T-cells and on a subset of thymocytes and NK cells)
7. CD16 (present on NK cells and granulocytes)
8. CD56 (present on NK cells and subset of T lymphocytes; also present on myeloma cells)
9. TIA-1 (cytotoxic granule-associated RNA binding protein), granzyme B, and perforin (proteins released by cytotoxic T-cells, inducing apoptosis) are expressed in cytotoxic T-cells and NK cells
G. Flow cytometry can easily detect clonality in B-cell malignancies (normal κ to λ light chain ratio ranges between 1:1 and 4:1). Clonality for T-cell malignancies can only be detected by molecular or cytogenetic studies.
H. Southern blot analysis is considered the gold standard for identifying clonal immunoglobulin heavy chain (IgH) gene or TCR gene rearrangements.
T lymphocytes normally rearrange four different TCR genes (TCRα, β, γ, and δ) to encode a unique antigen receptor expressed on their surface, and Southern blot analysis targets the TCRβ gene. In contrast to normal tissue, where only one band corresponding to the germline configuration is visible with this technique, both B-cell and T-cell malignancies will yield an additional band corresponding to the clonal population.
I. PCR. Because Southern blot analysis is labor intensive and expensive to perform, many laboratories have turned to PCR amplification methods to detect clonal IgH and TCR gene rearrangements.
The 200 variable segments of the IgH gene contain three highly variable and mutation-prone regions called complementary determining regions (CDR1, 2, and 3) that are interspersed between four conserved framework regions (FR1, 2, 3, and 4) that provide reliable targets for consensus primers. About 70% of B-cell malignancies harbor clonal rearrangements that are detectable with framework 3 primers, and about 15% to 20% of additional clonal rearrangements can be detected when framework 2 primers are used. Therefore, most laboratories use primers targeting frameworks 2 and 3 to detect at least 80% of all B-cell lymphomas.
The rearranged TCRγ gene is most suitable for clonal detection by PCR amplification in T-cell malignancies, because most T lymphocytes harbor rearrangements in 1 of the 11γ segments. Because many of these segments are homologous to one another, they can be targeted by a single consensus primer set. TCRδ and TCRα cannot be targeted because TCRδ is deleted during TCRα gene rearrangement and TCRα has such a diversity of possible rearrangements that these cannot be covered by current probe technology.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Lymph Nodes
Lymph Nodes
Anjum Hassan
Friederike Kreisel
