Fig. 19.1
A scanning electron micrograph of the lower basal turn of a human cochlea. The arrow indicates the direction of insertion of a cochlear implant electrode array into the scala tympani between the scala tympani fluid space and the spiral ganglion neurons located within Rosenthal’s canal is a thin sheet of mesothelium covering the trabeculated bony columns that guide the peripheral axons of these auditory ganglion cells to the sensory hair cells within the organ of Corti. Bar = 200 μm
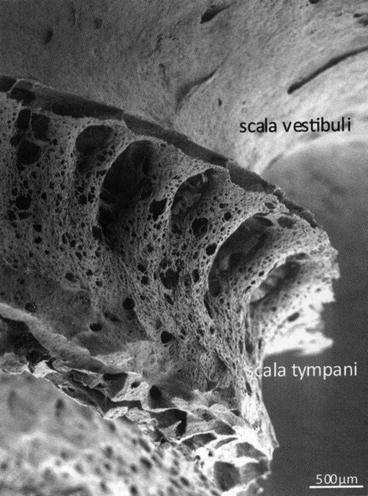
Fig. 19.2
A scanning electron micrograph of the thin trabeculated bony columns that enclose the spiral ganglion neurons that are located within Rosenthal’s canal. These are fragile structures easily damaged and also this micrograph depicts the routes of communication between the fluid space of the scala tympani and Rosenthal’s canal. Bar = 500 μm
As reviewed in Chap. 7 by Altschuler, sound pressure waves enter the ear canal and initiate vibrations of the tympanic membrane that are amplified by the ossicular chain of the middle ear. These vibrations travel to the oval window and create fluid shifts that travel through the cochlear partition to vibrate the basilar membrane in a tonotopic manner. The stereocilia of the auditory HCs are then displaced from their resting position, leading to inner hair cell (IHC) membrane depolarization, glutamate exocytosis into the IHC—afferent nerve terminal synaptic clefts, and electrical signaling to the auditory cortex through SG neuron activation. The outer hair cells (OHCs) are the mechanosensory receptor cells of the cochlea that are essential for detecting and amplifying mechanical vibrations of the basilar membrane for normal hearing. Fortunately, CIs can restore hearing when there has been a significant loss of the auditory OHCs and/or IHCs. Electrical signals from a CI bypass the need for these auditory HCs as it directly stimulates the SG neurons in a frequency-specific manner for auditory perception.
Placement of this electrode array into the scala tympani can be physically traumatic in even the most skilled hands and can induce intracochlear expression of various pro-inflammatory and pro-apoptotic factors that can lead to apoptosis of some or even all of the remaining HCs in the cochlea. By preventing programmed cell death of these mechanosensory cells of the OC following implantation, preservation of residual hearing can be achieved postoperatively, which has assumed increasing importance since the current trend is to implant these patients with hybrid devices that combine electrical and acoustic stimulation (Turner et al. 2004). Patients that have successfully preserved residual hearing after cochlear implantation can utilize both electric (i.e., from CI stimulation) and acoustic hearing (i.e., from amplified sound wave stimulation of functional HCs in the low-frequency region of the organ of Corti). Acoustic low-frequency hearing may allow patients to better distinguish fine pitch differences that should allow improved speech recognition in noise, melody recognition, and music appreciation (Turner et al. 2004; Gantz et al. 2005, 2006, 2009; Gfeller et al. 2006; Adunka et al. 2010, 2013; Carlson et al. 2011; D’Elia et al. 2012; Cosetti et al. 2013; Santa Maria et al. 2013).
Although low-frequency preoperative hearing thresholds may be preserved shortly after surgery, the auditory HCs may continue to degenerate with time and lead to long-term progressive loss of residual hearing (Santa Maria et al. 2013). This phenomenon may represent a progressive degeneration that originates from the patient’s underlying disease process, a prolonged low-level inflammatory process stemming from a foreign body reaction to the electrode array, or very likely a combination of both of these processes. Therefore, it is imperative to understand how cochlear implantation affects apoptosis and non-apoptotic initiated loss of auditory HCs to allow identification of solutions to prevent these processes, to preserve residual hearing long term and improve CI benefits after surgery and in the long term.
Another issue that leads to poor hearing outcomes following cochlear implantation is an increase in electrode impedance secondary to the development of local fibrosis and osteoneogenesis. Wound healing is a complex process that involves the expression of various pro-inflammatory cytokines, cross-communication across many cell types, and modulation by surrounding extracellular and vascular components. By understanding the sequential and cyclic events that occur on a molecular level to transform electrode insertion-initiated inflammation into scar tissue, drugs may be developed to target key events in this inflammatory cascade to slow or halt the progression of cochlear fibrosis and improve post-implantation speech and music perception.
19.2 Cochlear Implantation and Trauma-Induced Loss of Residual Auditory Hair Cells
As reviewed in Chap. 2 by Leeuwenburgh, apoptosis refers to a highly regulated form of programmed cell death that is characterized by cell blebbing and shrinkage followed by chromatin condensation and DNA fragmentation (Hengartner 2000). Apoptosis can occur through two main cell death signaling pathways: the intrinsic pathway (i.e., mitochondrial death pathway) and the extrinsic pathway [i.e., death receptor (DR) pathway]. Both pathways can independently activate downstream events that lead to apoptosis (caspase-dependent cell death) or apoptosis-like cell death [apoptosis-inducing factor (AIF) or endonuclease G (Endo-G)-mediated cell death]; however, cross-communication can also occur between these two signaling cascades and promote programmed cell death.
In the intrinsic pathway, stress signals, such as oxidative stress from expression of high levels of ROS, can initiate depolarization of the mitochondrial membrane to release intramembranous pro-death proteins into the cytoplasm of an affected cell. These mitochondrial proteins include cytochrome C (cyt. C), second mitochondria-derived activator of caspases (Smac)/direct inhibitor of apoptosis-binding protein with low pI (DIABLO), mammalian homolog of bacterial high temperature requirement protein (Omi/HtrA2), AIF, and Endo-G, cyt. C, Smac, and Omi/HtrA2 promote caspase-dependent activation of apoptosis, while AIF and Endo-G initiate caspase-independent pathways of cell death.
Cyt. C will bind to apoptotic peptidase-activating factor 1 (Apaf-1) and initiate the formation of an apoptosome, which is an oligomeric complex that can activate caspase-9 (an initiator caspase) to in turn initiate the activation of downstream caspases such as caspase-3 and -7 (downstream executor caspases) to promote cell death (Li et al. 1997). Smac, on the other hand, will bind to X-linked inhibitor of apoptosis proteins (XIAP), thereby disrupting XIAP–caspase interactions and releasing basal inhibition of caspases (e.g., caspases-3, -7, and -9) (Srinivasula et al. 2000). Similar to Smac, Omi/HtrA2 can bind and neutralize inhibitors of apoptosis (IAPs) to promote caspase-dependent apoptosis; however, the effects of Omi/HtrA2 are irreversible through cleavage of IAPs (Yang et al. 2003). AIF and Endo-G do not depend on caspase activation, but rather translocate into the cell nucleus and promote DNA fragmentation and chromatin condensation resulting in caspase-independent apoptosis of the affected cell (Susin et al. 1999; Li et al. 2001).
The extrinsic pathway is also known as the DR pathway as it requires binding of a death ligand to its complementary receptor on the cell membrane to activate caspase-8 and downstream events that lead to apoptosis. Tumor necrosis factor receptor 1 (TNFR1) and Fas are the best characterized receptors of this DR-activated cell death pathway; however, other receptors can also activate this signaling cascade. These receptors include tumor necrosis factor (TNF)-like receptor apoptosis-mediating protein (TRAMP, or DR3), TNF-related apoptosis-inducing ligand receptor 1 (TRAIL-R1, or DR4), and TRAIL-R2 (DR5) (Itoh and Nagat 1993; Tartaglia et al. 1993; Bodmer et al. 1997; Griffith et al. 1999; Chaudhary et al. 1997). When the death ligand binds to its complementary DR, several intracellular death domains containing adaptors, such as Fas-associated death domain (FADD) and TNFR1-associated death domain (TRADD), are recruited along with caspase-8 to form a death-inducing signaling complex (DISC) (Tartaglia et al. 1993; Chinnaiyan et al. 1995; Hsu et al. 1995; Schneider-Brachert et al. 2004). DISC is important for activation of caspase-3 and caspase-dependent programmed cell death. In addition, the extrinsic pathway signaling can promote cleavage of BH3 interacting-domain (Bid) death agonist to its active truncated form, tBid. Active tBid can lead to Bcl-2 associated X protein (Bax)-initiated depolarization of the mitochondrial membrane, pore formation, and activation of the intrinsic pathway to promote apoptosis (Li et al. 1998).
Cell death can also occur through mechanisms other than apoptosis, i.e., necrosis. Necrosis is a form of cell death that was thought to be unorganized and is characterized by increased permeability of the cell membrane, cellular edema with eventual cell membrane rupture, and release of intracellular contents into the surrounding environment. Recently, necrosis has been renamed “regulated necrosis” because it has been found to depend on specific signaling factors that lead to this morphologic form of cell death (i.e., RIP1 and -3). Stimuli that may initiate necrosis include alkylation DNA damage, excitotoxins, and ligation of DRs (Galluzzi et al. 2012).
Cochlear implantation trauma likely causes auditory HC and residual hearing loss from a combination of apoptosis, apoptosis-like cell death and regulated necrosis; however, currently there are no studies at this time that evaluate the role of regulated necrosis in post-implant loss of a patient’s residual hearing or loss of hearing in an animal model of EIT-induced hearing loss.
Cochlear implantation surgery and EIT cause apoptosis of auditory HCs through activation of both the mitochondrial and DR signaling pathways. Several events during cochlear implantation are linked to apoptosis of auditory HCs, and they include: (1) acoustic and vibrational injury from drilling of the ossicles, labyrinth bone, and/or cochleostomies; (2) blood byproducts from inadvertent entrance of blood into the cochlea; (3) displacement of bone dust into the perilymphatic compartments of the inner ear; (4) mechanical injury to the auditory HCs from the insertion of the electrode array; (5) traumatic penetration of the basilar membrane resulting in alterations of endocochlear potential (EP) and a mixing of endolymph with perilymph; (6) fracture of osseous spiral lamina; (7) electrode tip dissection of the spiral ligament and stria vascularis important for cochlear homeostasis; (8) immediate or delayed foreign body reaction; and at times, (9) inflammatory response from bacterial infection acquired during the process of CI.
Attention to various steps of cochlear implantation can help prevent loss of residual auditory HCs and hearing loss. Soft surgical techniques have been described to reduce the trauma and inflammatory response that result in apoptosis, or apoptosis-like cell death or regulated necrosis of auditory HCs following cochlear implantation. In brief, cochlear implantation involves drilling of the mastoid through a retro-auricular incision and identification of the incus and lateral semicircular canals as initial landmarks to guide the drilling of the facial recess. Once the facial recess is drilled, the stapes and promontory are identified and the bony ledge obscuring visualization of the round window is drilled to improve the angle for the insertion of the electrode array. If a cochleostomy is required, drilling will proceed using a small bur bit anterior–inferior to the round window membrane. The CI electrode array is carefully inserted through an incision in the round window membrane or through a pre-drilled cochleostomy using micro-instruments employed and guided under high magnification of an operating microscope. Figure 19.3 illustrates basic human temporal bone anatomy important for safe cochlear implantation.
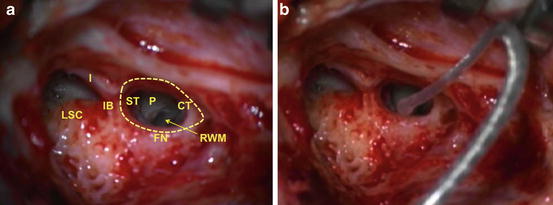
Fig. 19.3
Surgical anatomy for electrode insertion into a right cochlea. (a) A right mastoidectomy has been drilled and the lateral semicircular canal (LSC) and incus (I) are identified. These landmarks assist in identifying the incus buttress (IB), facial nerve (FN), and chorda tympani (CT) and safe drilling of the facial recess (dotted line). Once the facial recess is drilled, the stapedial tendon (ST) and the bony promontory (P) of the cochlea are visualized. Drilling of the promontory may be necessary to expose the round window membrane (RWM) for successful cochlear implantation or proper drilling of a cochleostomy anterior–inferior to the RWM. (b) A cochlear implant electrode is seen inserted through the RWM into the cochlea
Techniques of soft surgery in cochlear implantation aim to preserve the survival of auditory HCs important for residual acoustic hearing and are described below (Lehnhart 1993; Cohen 1997; Kiefer et al. 2004; Roberson 2005; Roland et al. 2005; Eshraghi 2006; Skarzynski et al. 2007; Berrettini et al. 2008; Jia et al. 2011; Postelmans et al. 2011; Havenith et al. 2013). The basis of this technique has scientific merit in preventing loss of auditory HCs and hearing loss following cochlear implantation as outlined and highlighted in detail in Fig. 19.4.
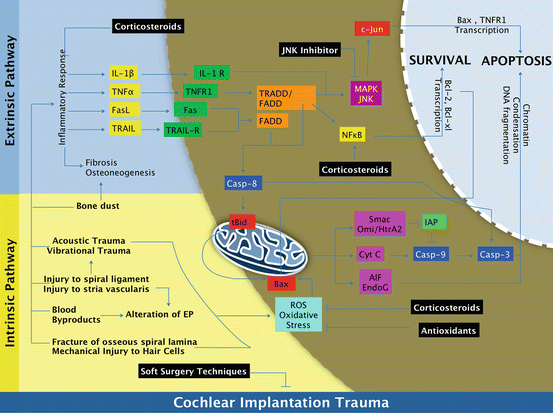
Fig. 19.4
Cochlear implantation and apoptosis. Cochlear implantation can result in acoustic and vibrational trauma, injury to the spiral ligament and stria vascularis, introduction of blood by-products into the cochlea, fracture of the osseous spiral lamina, mechanical injury to auditory hair cells, and alterations in the endocochlear potential (EP) that trigger expression of reactive oxygen species (ROS) and oxidative stress that promote the mitochondrial (or intrinsic) pathway of apoptosis (bottom half of diagram). The intrinsic pathway involves activation of Bax protein and release of mitochondrial proteins that promote apoptosis. Smac, Omi/HtrA2, and Cyt C initiate caspase-dependent apoptosis through activation of caspase-9 and -3, while AIF and Endo-G do not depend on caspases and translocate to the nucleus to initiate chromatin condensation and DNA fragmentation for apoptosis to occur. Cochlear implantation also initiates an inflammatory response and expression of several pro-inflammatory and pro-death cytokines, particularly IL-1β and TNFα. These cytokines trigger a signaling cascade that preferentially activates MAPK/JNK signaling and caspase-8 and -3 that promote the death receptor (or extrinsic) pathway of apoptosis (top half of the diagram). NFκB may also be activated in attempt for cell survival, but to a lesser extent. Caspase-8 from the extrinsic pathway can trigger apoptosis through the intrinsic pathway through tBid activation of Bax activity. Soft surgical techniques, corticosteroids, JNK inhibitors, and antioxidants can inhibit pro-death signaling pathways and promote cell survival pathways for auditory hair cell survival
Vibrations from drilling of the ossicular chain or on the bony promontory can be transmitted to the inner ear and injure the stria vascularis and auditory HCs. The exact mechanism of vibration-induced inner ear damage is not entirely clear but may involve alterations of the EP via injury to the intermediate cells of the stria and increased permeability of the stria vascularis (Seki et al. 2001; Marcus et al. 2002; Hashimoto et al. 2006). Changes in EP likely affect the homeostasis of auditory HCs leading to activation of the mitochondrial pathway of apoptosis or apoptosis-like cell death, since maintenance of the EP is crucial for the survival of the OHCs (Takeuchi et al. 2000; Teubner et al. 2003; Nouvian et al. 2003; Wangemann et al. 2004). Vibration is also associated with intracochlear expression of TNFα and TNFR1, which together can initiate the DR pathway and downstream signaling that leads to apoptosis of auditory HCs (Zou et al. 2005; Dinh et al. 2008a, b; Haake et al. 2009). Vibrational injury can affect HC viability from the apical to the basal turns of the cochlea and initially affects the outer most rows of OHCs before proceeding to injure the second and then first rows of OHCs located toward the cochlear modiolus. SG neuron density was also reduced following exposure to vibration trauma (Bochnia et al. 2005). Furthermore, vibrations from drilling can initiate hearing threshold shifts in both laboratory animals and humans (Zou et al. 2001; Karatas et al. 2007; Migirov and Wolf 2009; Moussavi-Najarkola et al. 2012). High-frequency vibrations have been shown to be more detrimental to hearing than low-frequency vibrations with older animals more susceptible to vibration-induced threshold shifts than younger animals, and surgical drilling with steel cutting burs more likely to affect otoacoustic emissions than drilling performed with diamond burs (Zou et al. 2001; Goyal et al. 2013).
Acoustic trauma represents another mode of injury to auditory HCs during cochlear implantation. Sound emissions from drilling near the cochlea can travel to the sensory receptor tissues through the oval and round windows. Different surgical drilling burs produce various levels of sound intensity with steel cutting burs producing higher sound pressure levels than diamond burs (Dalchow et al. 2013). Loud noise exposures can injure the stria vascularis, leading to changes in the potassium concentration of the endolymph, alterations in the EP, and damaging levels of oxidative stress (Syka et al. 1981; Wang et al. 1992; Poje et al. 1995; Yamane et al. 1995; Ohlemiller et al. 1999; Ohinata et al. 2000; Hirose and Liberman 2003). The mitochondrial cell death pathways are then triggered, resulting in release of cyt. C and caspase-3-dependent cell death of auditory HCs and/or release of Endo-G and AIF responsible for DNA fragmentation and caspase-independent apoptosis (Henderson et al. 2006; Hu et al. 2002; Nicotera et al. 2003; Yamashita et al. 2004; Han et al. 2006). The DR pathway is also activated following acoustic trauma through expression of TNFα and the TNF superfamily of receptors and downstream signaling that also involves activation of the pro-apoptotic mitogen-activated protein kinase (MAPK) signal cascade which can involve activation of the c-Jun N-terminal kinase (JNK) pathway as well as caspase-8-dependent events of programmed cell death (Pirvola et al. 2000; Nicotera et al. 2003; Wang et al. 2003; Hu et al. 2009; Jamesdaniel et al. 2011). In addition, noise-induced injury to the inner ear can cause an acute loss of afferent nerve terminals and delayed degeneration of the cochlear nerve (Kujawa and Liberman 2009). Thus, it is vital to limit the effects of both acoustic and vibration-induced trauma to the cochlea during the process of CI in order to reduce intracochlear apoptotic and apoptotic-like signaling that can interfere with conservation of residual hearing during and after cochlear implantation.
Surgical techniques to minimize bone particles and blood from entering the cochlea will likely reduce the inflammatory burden in the cochlea that can lead to loss of auditory HCs after electrode array insertion. Though there is no clear evidence that bone pâté in the cochlea will cause loss of residual hearing, it can promote osteoneogenesis and fibrosis within the scala tympani that may cause inference with electrode impedance and a delayed electrode array extrusion response (Clark et al. 1995; McElveen et al. 1995). On the other hand, intrascalar blood can lead to temporary and permanent threshold shifts particularly in the apical region when autologous blood is injected into the cochlea of a guinea pig (Radeloff et al. 2007). The hearing threshold shifts are likely a result of erythrocyte degradation which leads to recruitment of inflammatory cytokines (e.g., TNFα, IL-1β, IL-6) and inflammatory enzyme expression [e.g., matrix metalloproteinase-9 (MMP-9) and MAPK], production of ferrous ions leading to excessive levels of ROS production, and changes in EP from potassium spillage into the scala tympani (Van Bergen et al. 1999; Mathiesen et al. 1997; Maddahi et al. 2012). These apoptotic stimuli will activate both intrinsic and extrinsic pathways of apoptosis and apoptosis-like cell death in auditory HCs, thus stressing the importance of avoiding the introduction of blood into the cochlea during the process of CI.
Conservative use of sodium hyaluronate gel during cochlear implantation may help protect residual hearing. Sodium hyaluronate is injected over the endosteum with a micro-lancet prior to penetrating the membrane, but after the bone has been drilled away from the cochleostomy site. This prevents entrance of blood and bone pâté into the cochlea and prevents perilymph from escaping from the scala tympani prior to electrode array insertion (Laszig et al. 2002). The gel also acts as a lubricant to aid smooth insertion of the electrode array. However, excessive amounts of intracochlear sodium hyaluronate gel can cause auditory HC injury and severe sensorineural hearing loss (Roland et al. 2005). The mechanism of injury is unclear and may involve drug toxicity or mechanical injury from injection that initiates oxidative stress and programmed cell death of auditory HCs. There is no evidence that sodium hyaluronate use alone prevents threshold shifts following cochlear implantation, but it is associated with residual hearing preservation when combined with other soft surgical techniques (Skarzynski et al. 2002; Kiefer et al. 2004; Balkany et al. 2006; Fraysse et al. 2006; Garcia-Ibanez et al. 2009). In addition, suctioning of the perilymph should be avoided during this time to prevent rapid fluid shifts and changes in the endocochlear potential that can lead to oxidative stress and auditory HC programmed cell death; the use of sodium hyaluronate may also act as a small buffer when pericochlear suctioning is unintentional.
The characteristics of an electrode array (i.e., stiff vs. flexible, thin vs. thick, straight vs. contoured), the position of the cochleostomy, the depth of insertion, and their contribution to soft surgical techniques are extremely important. Appropriate selections will aid in smooth electrode insertion parallel to the trajectory of the basal turn, thereby avoiding inadvertent mechanical injury to the auditory HCs, basilar membrane penetration and alteration of EP, fracture of osseous spiral lamina, injury of SG neurons, and dissection of spiral ligament and stria vascularis (Kiefer et al. 2004; Briggs et al. 2005; Gantz et al. 2005; Gstöettner et al. 2006, 2008; Adunka et al. 2007; Baumgartner et al. 2007; Li et al. 2007a, b; Berrettini et al. 2008; Bruce et al. 2011; Helbig et al. 2011a, b; Eshraghi et al. 2012). In addition, intracochlear injuries can lead to osteoneogenesis and fibrosis that can affect electrode impedance (Li et al. 2007a, b; Fayad et al. 2009). The culmination of EIT will not only activate a very robust inflammatory reaction but also result in activation of both the mitochondrial and DR-mediated apoptosis and apoptosis-like pathways to initiate loss of auditory HCs.
Oxidative stress and ROS production from EIT-associated mechanical disruption of intracochlear structures and fluxes in EP can initiate the intrinsic pathway. The mitochondria will then release pro-apoptotic mitochondrial proteins that lead to caspase-3-dependent cell death and Endo-G initiated nuclear condensation, DNA fragmentation, and caspase-independent cell death (Bas et al. 2012; Eshraghi et al. 2013). Expression of pro-inflammatory cytokines (i.e., TNFα and IL-1β) and pro-inflammatory enzymes [inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2)] can also promote production of high levels of ROS and intrinsic pattern of apoptosis (Friedlander et al. 1996; Ho et al. 1998; Nam 2006). TNFα, particularly, can bind to its receptor TNFR1 to initiate DR-associated apoptosis of auditory HCs through activation of MAPK and JNK signaling pathways initiating an upregulation in the expression of pro-apoptotic Bax gene and protein expression, upregulation of TNFR1 gene for apoptosis propagation, downregulation of pro-survival Bcl–2 and Bcl–xl gene expressions, and caspase activity (Dinh et al. 2008a; Eshraghi et al. 2013). As demonstrated, expression of these factors can promote downstream pathways that act cumulatively to tilt the pendulum from cell survival to cell death, resulting in a progressive loss of auditory HCs and residual hearing at several days post-EIT following a cochlear implantation (Eshraghi et al. 2005). Several therapeutic agents that target particular aspects of the EIT-induced cell death pathways in auditory HCs have demonstrated protection of residual hearing following an EIT event (see Sect. 20.4, below).
19.3 Cochlear Implantation, Inflammation, and Fibrosis Signaling
Cochlear fibrosis and ossification can occur as a result of CI-EIT-initiated inflammatory cascade and can affect efficiency of the CI (Fig. 19.5). Higher levels of current would be necessary to overcome the impedance caused by scar tissue and bone growth (Choi and Oghalai 2005). Targeting the mediators responsible for fibrosis and neo-osteoneogenesis after a CI-EIT insult may improve hearing outcomes following cochlear implantation by reducing intracochlear scar tissue and new bone formation. In the case of device failure, CI reimplantation will be less risky if the cochlear lumen is not obstructed by fibrous tissue and newly formed bone.
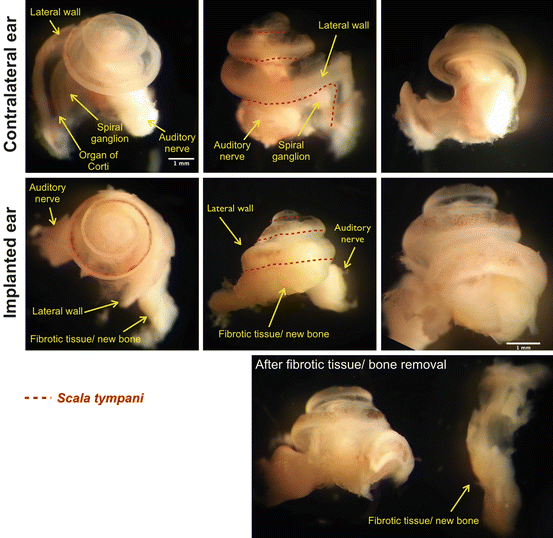
Fig. 19.5
An increase in impedance is well correlated with an increase of fibrotic tissue growth in the scala tympani located in the area of the inserted electrode array. The macroscopic images correspond to a contralateral (not implanted) control cochlea and an implanted cochlea of an adult Norway brown rat. Fibrotic and/or bone tissue are found along the scala tympani, where the electrode insertion trauma occurred. The scala tympani for the three turns of the rat cochlea is located under the red dashed line. Bar = 1 mm
The inflammatory response that occurs following CI-EIT is a complex biological process that involves vascular and cellular components as well as soluble substances.
Disruption of blood vessels and leakage of blood components into surrounding tissue can lead to aggregation of platelet cells and a signaling cascade that produces a clot rich in fibrin and other extracellular matrix proteins (Bergmeier and Hynes 2012). Soon after, fibrin is degraded, several enzymatic proteins called the complement system are activated, and various cytokines and chemokines are released locally. This constellation of events leads to the recruitment of immune cells to the wound site and the initiation of the inflammatory phase of wound healing.
Neutrophils, a type of white blood cell, are the first immune cells to arrive following activation of the inflammatory response. They are a major source of pro-inflammatory cytokines, such as interleukin (IL)-1α, IL-1β, IL-6, and TNFα (Mutsaers et al. 1997). Monocytes, another type of immune cell, are attracted to the wound by local expression of chemokines. Exposure to these cytokines released by neutrophils can cause monocytes to differentiate into M1 macrophages, which secrete other cytokines. M1 macrophages also stimulate inducible nitrous oxide synthase (iNOS) activity and expression of ROS and reactive nitrogen species (RNS) that can be detrimental to surrounding cells. Monocytes that reach the clot extracellular matrix proteins can be stimulated by other cytokines (e.g., IL-4, IL-10, and IL-13) and develop into M2 macrophages. M1 macrophages are involved in clearance of injured cells, while M2 macrophages release anti-inflammatory cytokines and promote wound healing and tissue repair (Delavary et al. 2011; Airel and Timor 2013).
M2 macrophages release high levels of growth factors from the TGF-β family of proteins, which activate a fibroproliferative response that leads to wound healing. There are three isoforms of TGF-β proteins that participate in wound healing. TGF-β1 is associated with fibrocyte proliferation, fibrosis, and osteoneogenesis while TGF-β2 and TGF-β3 are important for scarless wound healing (Li et al. 2006).
In an in vitro model of EIT, there was an early upregulation of gene transcripts of pro-inflammatory cytokines TNF-α and IL-1β in the apical turns of rat OC explants 24 h post-EIT. At a later time point (i.e., 96 h), there was a dramatic downregulation of TNF-α and IL-1β and a significant increase in TGF-β1 gene expression levels (Bas et al. 2012). These changes occur to a lesser extent in the middle and basal turns of these EIT-OC explants. Early expression of TNF-α and IL-1β represent the initial inflammatory response that occurs following EIT and late expression of TGF-β1 is largely responsible for the fibrosis that ensues.
In the same model, increases in TGF-β3 were demonstrated in the middle and basal turns of rat OC explants following EIT, and these levels were magnified by Dexamethasone (DXM) treatment. TGF-β3 plays an important role in scarless wound healing and this increase in TGF-β3 in the middle and basal turn segments may be beneficial in reducing the inflammatory process and encouraging healing at the trauma site with lower levels of fibrotic scar tissue formation (Tandon et al. 2010). These results also suggest that treatment with DXM may encourage wound healing and tissue repair with less fibrosis.
Wnts are a family of proteins that bind and activate serpentine membrane receptors encoded by the Frizzled gene family. There are three main Wnt signaling pathways which include: (1) the Wnt/β-catenin (canonical) pathway; (2) the planar cell polarity (non-canonical) pathway; and (3) the Wnt/Ca2+ (non-canonical) pathway. The differential expression of various Wnt proteins and signaling cascades during embryogenesis is involved in cochlear differentiation (Daudet et al. 2002). Both Wnt and TGF-β proteins are important growth factors in the regulation of tissue homeostasis, development, differentiation of mesenchymal stem cells, regulation of mesenchymal cell function, fibroblast proliferation, and wound healing (Fig. 19.6) (Colwell et al. 2006; Jian et al. 2006; Salazar et al. 2009).
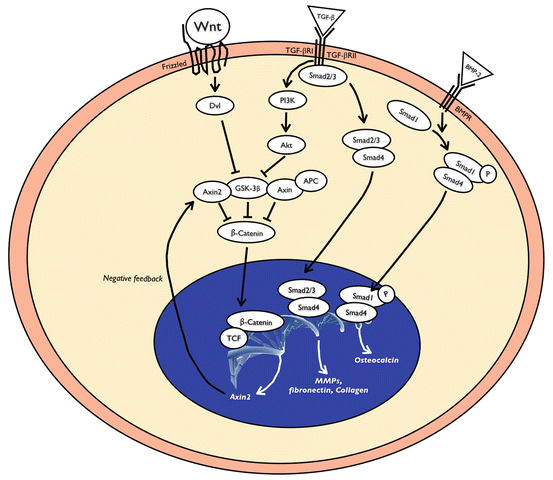
Fig. 19.6
TGF-β binds to its heteromeric receptor complex of type I and II and the intracellular signal is propagated downstream through Smads into the nucleus. Wnt signaling is initiated by secreted Wnt proteins, which bind to the Frizzled family of receptors. The activation of the receptor leads to the inhibition of GSK-3β-dependent phosphorylation of β-catenin and the stabilization and accumulation of cytosolic β-catenin. The accumulated β-catenin binds TCF/LEF transcription factors and activates transcription of target genes that will lead to the fibrotic tissue growth. TGF-β may cooperate with Wnt/β-catenin signaling. For example, TGF-β is known to activate PI3K which inhibits GSK-3β and could result in an increase of cytosolic β-catenin levels. BMP-2 binds its receptor and phosphorylates SMAD-1 and -5, which translocate into the nucleus and regulate the transcription of target genes, such as osteocalcin which is involved in bone formation
Wnt proteins promote the differentiation of fibroblasts into myofibroblasts, which express α-smooth muscle actin (α-SMA) (Akhmetshina et al. 2012). Myofibroblasts produce an extracellular matrix rich in collagen-1 and laminin and play a key role in fibrotic diseases. Cells of the inner sulcus of the cochlea demonstrate increased levels of α-SMA following EIT, which suggest an epithelial to mesenchymal cell transition (EMT) of these cells (Van De Water et al. unpublished data). These EMT cells may play an important role in the production of stress fibers and collagen type 1A in the lateral wall tissues and intracochlear fibrosis.
Bone morphogenic proteins (BMPs) are members of the TGF-β gene superfamily, which signal through Smad transcription factors. Interactions between BMPs and the Wnt/β-catenin pathway are crucial for osteoneogenesis (Fig. 19.6) (Chen et al. 2007; Chen and Alman 2009). Activation of these proteins may be responsible for new bone formation in the cochlea after CI-EIT.
In summary, CI-EIT can initiate an inflammatory process that leads to fibrosis and osteoneogenesis within the cochlea that can interfere with CI impedances and ease of CI reimplantation. The electrode array can also produce a foreign body reaction resulting in a chronic inflammatory condition that perpetuates scar and bone formation.
19.4 Drug Therapies to Prevent or Lessen Cochlear Implantation Trauma-Induced Sensory Cell and Hearing Losses
Corticosteroids and inhibitors of JNK signaling are two main drug classes that have been investigated for the treatment against CI-EIT-induced HC and hearing losses. The molecular mechanisms behind otoprotection by these two agents have been studied in depth and demonstrate consistent results among several studies.
DXM is a potent synthetic corticosteroid that demonstrates strong anti-inflammatory and immunosuppressant effects. It is used routinely in the treatment of various inner ear disorders to reduce the inflammatory reactions that may promote death of auditory HCs and hearing loss following an insult to the cochlea. There are consistent results across several experimental studies in animals that have demonstrated the effectiveness of DXM treatment in preventing auditory HC death and hearing loss following an EIT event.
TNFα is a cytokine released during EIT and can activate the DR pathway of apoptosis and cross-communicate through tBid to activate mitochondrial death signaling; as a result, TNFα is used as an apoptotic stimulus to mimic an environment of EIT to test the effectiveness of DXM in protecting auditory HCs from EIT-induced loss. DXM treatment of TNFα-injured OC explants in vitro demonstrated significantly higher levels of auditory HC survival when compared to explants damaged by TNFα without treatment (Dinh et al. 2008a). The result of several inhibitor studies conclude that DXM otoprotection is initiated when DXM traverses the cell membrane and binds to its glucocorticoid receptor (GCR) in the cytoplasm and activates pro-survival phosphatidylinositol 3-kinase (PI3K), Akt (also called protein kinase B, PKB), and nuclear factor kappa B (NFκB) proteins that lead to increased transcription of Bcl–2 and Bcl–xl anti-apoptosis genes along with the downregulation of Bax and TNFR1 gene expression levels resulting in an inhibition of caspase-dependent apoptosis (Haake et al. 2009; Dinh et al. 2008a). Similar mechanisms of action were demonstrated using polymer-eluted DXM against TNFα-initiated loss of auditory HC in vitro. DXM eluted from a biorelease polymer showed preservation of IHCs and OHCs that were comparable to levels of these sensory receptor cells present in control OC explants (Dinh et al. 2008b). Eluted DXM also mitigated expression of Bax and TNFR1 genes associated with TNFα ototoxicity while also increasing expression of pro-survival Bcl–2 and Bcl–xl in rat OC explants in vitro (Dinh et al. 2008b).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


