resting, when the channel is closed but able to open in response to a change in transmembrane potential,
 open, when the channel opens in response to an action potential and allows the rapid influx of Na+ ions through to the cytoplasm,
open, when the channel opens in response to an action potential and allows the rapid influx of Na+ ions through to the cytoplasm, inactivated due to a very rapid change in conformation at the cytoplasmic end of the channel that occurs very soon after the action potential has passed. During this stage the channel is resistant to depolarising influences, but sensitivity returns when the membrane potential is restored to the resting level.
inactivated due to a very rapid change in conformation at the cytoplasmic end of the channel that occurs very soon after the action potential has passed. During this stage the channel is resistant to depolarising influences, but sensitivity returns when the membrane potential is restored to the resting level.There is considerable redundancy in the membrane Na+ channels; as a consequence, nerve conduction can continue even when 90% of the channels are blocked.
Local anaesthetics block the voltage-dependent Na+ channels that depolarise the cell. They bind to the Na+ channel at a site on the inner surface of the membrane and hold them in an inactivated state. Local anaesthetics progressively interrupt Na+ channel-mediated depolarisation until conduction fails. The probability that propagation of a nerve impulse will fail at a particular segment of the nerve is related to:




In general, nerve transmission is blocked in smaller-diameter fibres before that in larger fibres (Table 18.1). The myelinated Aδ and small non-myelinated C fibres that transmit pain (nociceptive fibres) are blocked before larger sensory and motor fibres. Therefore, pain pathways are most rapidly and intensely blocked by local anaesthetics (Table 18.1), and also show the longest duration of local anaesthetic effect. In myelinated nerves the drug penetrates at the nodes of Ranvier and must block at least three consecutive nodes to produce conduction block. Unmyelinated nerves must be blocked over a sufficient length and around the full circumference of the nerve.
Table 18.1
Nerve fibres and their responsiveness to local anaesthetics
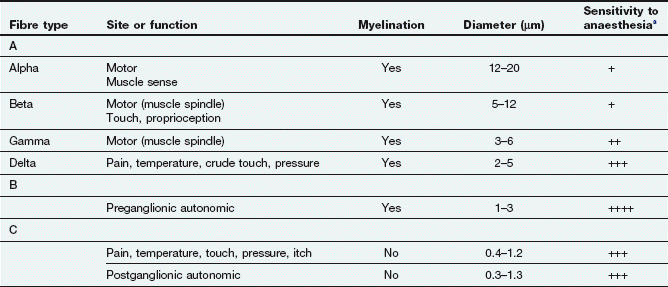
aIncreasing number of + indicates increasing sensitivity to local anaesthesia.
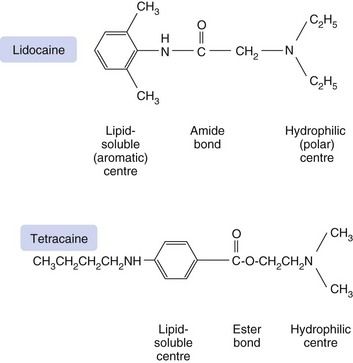
Structural requirements of local anaesthetics
The action of local anaesthetics results mainly from binding of the ionised form of the anaesthetic to a site on the inside (cytoplasmic opening) of the Na+ channel. Membrane penetration, however, is better in the non-ionised (lipid-soluble) form. The structural requirements for local anaesthetic activity appear to involve a minimum of a lipid-soluble hydrophobic aromatic ring structure connected to a hydrophilic amine group by a short ester or amide intermediate linkage (Fig. 18.1). Clinically used potent local anaesthetics are all secondary or tertiary amines with an intermediate amide or ester bond. The length of the intermediate bonding chain is critical to local anaesthetic activity and is optimal between three and seven carbon atoms or equivalent atoms. The lipophilic aromatic group enables the molecule to cross the nerve membrane, and the potency of the drug is directly related to its lipid solubility.
Specific intraneuronal binding
Local anaesthetics bind to a receptor protein within the cytoplasmic opening of the Na+ channel, and compounds with high protein-binding affinity stay at the site of action for longer and have a long duration of action. Thus, procaine, which has a low binding affinity (6% protein bound), has a very short duration of action, whereas bupivacaine has a high binding affinity (95% protein bound) and a long duration of action.
The pKa of the drug determines the extent of ionisation at physiological pH and the speed of onset of the conduction block. All local anaesthetics are weak bases and will be relatively more ionised at a pH below the pKa (which for most local anaesthetics is between 7.7 and 9.1). Because the water solubility of a local anaesthetic is greatest in the ionised form, injectable preparations are formulated as the hydrochloride salts with a pH of 5.0–6.0. However, the base (non-ionised) form is more lipid-soluble and more readily penetrates lipid membranes; therefore, after injection, the drug solution (pH 5.0–6.0) must be buffered in the tissues to physiological pH (7.4) before a significant amount of non-ionised local anaesthetic is available to penetrate the nerve and reach its site of action. A drug with a high pKa will be more ionised at physiological pH, and the speed of onset of anaesthesia will be slower. Alkalinisation of the injected solution by adding bicarbonate will increase the proportion of the drug in its non-ionised lipid-soluble form and therefore increase the rate of absorption of the anaesthetic into the nerve which will accelerate the onset of action.
In contrast, it is the ionised form of the drug that binds to the receptor within the cytoplasmic opening of the Na+ channel. Drugs with a higher pKa will re-ionise to a greater extent within the cell (pH 7.4) and produce more effective blockade. The majority of the local anaesthetic molecules therefore pass across the cell membrane in their non-ionised form and enter the cytosol where they become ionised and available to bind to the protein receptor via the cytoplasm. The binding site is most accessible when the channel is in its open (activated) state (Fig. 18.2). For this reason the effectiveness of most local anaesthetics is dependent on the frequency of firing of the neuron (use-dependency), and a faster onset of local anaesthesia occurs in rapidly firing neurons. Once the local anaesthetic has bound to the channel, the influx of Na+ is blocked and the channel remains in the inactivated state and resistant to further depolarisation. The local anaesthetic does not dissociate from the channel when it is inactivated as it has a high affinity for the channel in this state. The local anaesthetic leaves the binding site when the membrane potential returns to its resting level, and the channel is in the resting state. This is further facilitated when the cytoplasmic concentration of the drug decreases as it diffuses away from the site of administration.
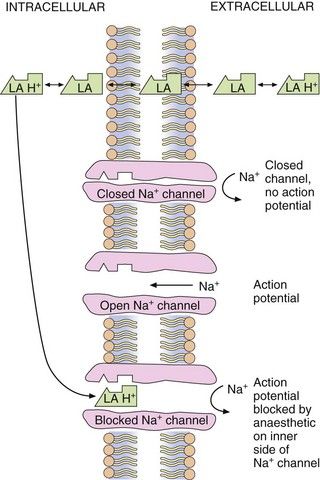
Fig. 18.2 Site and mechanism of action of local anaesthetics.
Local anaesthetics are weak bases and exist in an equilibrium between ionised (LA + H+) and non-ionised (LA) forms. The non-ionised form is lipid-soluble and crosses the axonal membrane, allowing the ionised form to bind to the intracellular end of the receptor. Some non-ionised anaesthetic may reach the ion channel by diffusion within the cell membrane, become ionised within the Na+ channel and then bind to the receptor site.
Local anaesthetics bind to K+ channels, but very weakly compared to Na+ channels. This binding may contribute to toxic effects of local anaesthetics in other organs such as the heart.
Local anaesthetics also bind to other ion channels and cell receptors, including presynaptic Ca2+ channels, tachykinin type 1 receptors, glutamate, bradykinin B2 and acetylcholine receptors. These actions may be involved in reducing nociceptive neurotransmission and in the production of spinal anaesthesia.
Pharmacokinetics
As seen above, the speed of onset of local anaesthetic action is largely determined by the physicochemical properties of the drug molecule. The duration of action of local anaesthetics is dependent on the degree of receptor binding (see above) and on their rate of removal from the site of administration, rather than their systemic elimination by metabolism. Most local anaesthetics cause vasodilation at the site of injection, which will enhance their removal. In contrast, cocaine, which blocks noradrenaline reuptake by noradrenergic neurons (Ch. 4), produces intense vasoconstriction. Because of the risk to local tissue integrity associated with the vasoconstriction, cocaine is never given by injection and its medical use is restricted to topical anaesthesia in otolaryngology. The duration of action of any local anaesthetic can be extended considerably by co-administration with a vasoconstrictor such as an α1-adrenoceptor agonist, for example adrenaline (epinephrine). However, the pH of the solution must be 2.0–3.0 to prevent decomposition of the adrenaline (epinephrine). Local anaesthetic preparations with other vasoconstrictors such as phenylephrine or felypressin are also available.
Once the local anaesthetic has diffused away from the site of administration it enters the general circulation and undergoes elimination from the body. Most local anaesthetics have an intermediate amide bond, and are eliminated at least in part by hepatic hydrolysis of the amide bond. The half-life of amide local anaesthetics within the circulation is generally between 1 and 3 h. In contrast, the plasma half-lives of the ester drugs procaine and tetracaine are 3 min or less, since ester bonds are very rapidly hydrolysed by plasma esterases.



