FIGURE 12-1. Basic structure of a liver lobule including the lymph flow system comprised of the spaces of Disse and interlobular lymphatics. (Reproduced with permission from Guyton AC. Medical Physiology. 5th ed. Philadelphia, PA: WB Saunders; 1976.)
The liver is a complex organ with a prominent role in all aspects of the body’s biochemistry. It takes up amino acids absorbed by the intestines, processes them, and synthesizes them into circulating proteins including albumin and clotting factors. The liver is also involved in the breakdown of excess amino acids and processing of byproducts including ammonia and urea. The liver plays a similar role in absorbing carbohydrates from the gut, storing them in the form of glycogen, and releasing them as needed to prevent hypoglycemia. Most lipid and lipoprotein metabolism, including cholesterol synthesis, occurs in the liver. The liver is the primary location for detoxification and excretion of a wide variety of endogenous substances produced by the body (including sex hormones) as well as exogenous substances absorbed by the intestines (including a panoply of drugs and toxins). Thus, in patients with liver failure, standard dosing of some medications can lead to dangerously high serum concentrations and toxicity. The role of the liver in bilirubin metabolism is explored further below.
With its double blood supply, large size, and critical role in regulating body metabolic pathways, the liver is affected by many systemic diseases. Although numerous illnesses affect the liver, it has tremendous reserve capacity and can often maintain its function in spite of significant disease. Furthermore, the liver is one of the few human organs capable of regeneration.
The pancreas is an elongated gland located in the retroperitoneum. Its head lies in close proximity to the duodenum, and the pancreatic ducts empty into the duodenum. The pancreas has both exocrine glands (which secrete digestive enzymes into the duodenum) and endocrine glands (which secrete hormones directly into the circulation).
The pancreatic exocrine glands produce enzymes that aid in digestion of proteins, fats, and carbohydrates (including trypsin, chymotrypsin, lipase, and amylase). Insufficient enzyme production (i.e., pancreatic exocrine insufficiency) is associated with malabsorption of nutrients, leading to progressive weight loss and severe diarrhea.
The pancreatic endocrine glands produce many hormones including insulin and glucagon. Insufficient insulin production leads to diabetes mellitus. Thus, the pancreas plays an important role in digestion and absorption of food as well as metabolism of sugar. Like the liver, the pancreas has a tremendous reserve capacity; over 90% glandular destruction is required before diabetes or pancreatic insufficiency develops.
AN INTRODUCTION TO LIVER TESTS AND THE LFT PANEL
Investigation of liver disease often begins with obtaining a panel of liver tests generally referred to as the LFT panel or LFTs (liver function tests). This panel may vary slightly between hospitals and labs but generally includes the aminotransferases, also referred to as transaminases, AST and ALT (which stand for aspartate aminotransferase and alanine aminotransferase, respectively), bilirubin, alkaline phosphatase (ALP), and albumin. The term liver function test is a misnomer because not all of these tests actually measure liver function (specifically, transaminases reflect liver injury). Additionally, the liver has several functions, and different tests reflect these different functions. Table 12-1 divides liver tests into rough categories. Although there is considerable overlap between these categories, these divisions may provide an initial framework for understanding the LFT panel.
ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma-glutamyl transpeptidase; INR = international normalized ratio; PT = prothrombin time.
This grouping of tests mirrors a division of liver diseases into two broad categories: cholestatic and hepatocellular. In cholestatic disease, there is an abnormality in the excretory function of the liver (i.e., namely secretion of bile by hepatocytes and passage of bile through the liver and bile ducts into the duodenum). In hepatocellular disease, there is primary inflammation and damage to the hepatocytes themselves (e.g., due to viral infection of the hepatocytes). These two categories may overlap because disease of the hepatocytes (hepatocellular processes), if severe enough, will also lead to derangement of bile secretion. However, the distinction between primarily cholestatic versus primarily hepatocellular diseases and in turn LFT patterns remains useful and fundamental.2 Perhaps further confusing this is that liver tests may be abnormal in patients with diseases that do not affect the liver.
The range of normal laboratory values used here is taken from Harrison’s Principles of Internal Medicine, 18th edition, and Nelson’s Textbook of Pediatrics, 19th edition. Reference ranges may vary slightly between different laboratories, and most laboratories will list their reference ranges along with lab results. Listed normal ranges are for adult patients, and pediatric patients will often have different ranges of normal values.
TESTS OF SYNTHETIC LIVER FUNCTION
As discussed above, one of the functions of the liver is to synthesize proteins that circulate in the blood, including albumin and clotting proteins. Measurement of the levels of these proteins in the blood provides a direct reflection of the ability of the liver to synthesize them. The liver has an enormous reserve function, so that it may synthesize normal amounts of proteins despite significant liver damage. Therefore, tests of synthetic function are not sensitive to low levels of liver damage or dysfunction. Inadequate protein synthetic function is mainly limited to hepatic cirrhosis, scarring of the liver that can result from years of alcohol abuse, inflammation, or massive liver damage (e.g., due to alcoholic liver disease, severe acute viral hepatitis, unrecognized and untreated chronic hepatitis, or potentially lethal toxin ingestion). In these situations, measuring synthetic function may be useful in determining prognosis by reflecting the degree of hepatic failure. The most commonly used tests of protein synthetic function are albumin and prothrombin time (PT).
Albumin
Normal range3: 4.0–5.0 g/dL
Albumin is a major plasma protein that is involved in maintaining plasma oncotic pressure and the binding and transport of numerous hormones, anions, drugs, and fatty acids.4 The normal serum half-life of albumin is about 20 days, with about 4% degraded daily.5 Because of albumin’s long half-life, serum albumin measurements are slow to fall after the onset of hepatic dysfunction (e.g., complete cessation of albumin production results in only a 25% decrease in serum concentrations after 8 days).1 For this reason, levels are often normal in acute viral hepatitis or drug-related hepatotoxicity.2 Alternatively, albumin is commonly reduced in patients with chronic synthetic dysfunction due to cirrhosis.
Albumin levels may be low due to a variety of other abnormalities in protein synthesis, distribution, and excretion in addition to liver dysfunction. These include malnutrition/malabsorption, protein loss from the gut, kidney, or skin (as in nephrotic syndrome, protein-losing enteropathy, or severe burns, respectively), or increased blood volume (e.g., following administration of large volumes of intravenous [IV] fluids).6–9 Albumin is a negative acute phase reactant meaning that in the setting of systemic inflammation (e.g., as due to infection or malignancy), the liver will produce less albumin. Severely ill hospitalized patients commonly have low albumin levels due to a combination of poor nutrition, systemic inflammation, and IV fluid administration. Extremely low albumin concentrations carry a poor prognosis for this reason irrespective of any particular liver disease. Given the numerous causes of a low albumin level, it is important to interpret it within the context of each patient. For example, in a patient with metastatic cancer and no known liver disease, a low albumin level suggests decreased nutritional intake and advanced malignancy with systemic inflammation. Alternatively, in a patient with known cirrhosis a low albumin level suggests severe chronic liver failure. In a patient with no known medical disease, a low albumin level suggests the presence of significant disease and requires further investigation.
Hypoalbuminemia itself is usually not associated with specific symptoms or findings until concentrations become quite low. At very low concentrations (<2–2.5 g/dL), patients can develop peripheral edema, ascites, or pulmonary edema. Albumin normally generates oncotic pressure, which holds fluid in the vasculature. Under conditions of low albumin, fluid leaks from the vasculature into the interstitial spaces of subcutaneous tissues or into the body cavities. Finally, low albumin concentrations affect the interpretation of total serum calcium and concentrations of drugs that are highly protein bound (e.g., phenytoin and salicylates).
Hyperalbuminemia is seen in patients with marked dehydration (which concentrates their plasma), where it is associated with concurrent elevations in blood urea nitrogen (BUN) and hematocrit. Patients taking anabolic steroids may demonstrate truly increased albumin concentrations, but those on heparin or ampicillin may have falsely elevated results with some assays. Hyperalbuminemia is asymptomatic.
Prealbumin (Transthyretin)
Normal range3: 17–34 mg/dL
Prealbumin is similar to albumin in several respects: it is synthesized primarily by the liver; it is involved in the binding and transport of various solutes (thyroxin and retinol); and it is affected by similar factors that affect albumin levels. The primary difference between the two proteins is that prealbumin has a short half-life (2 days, compared to 20 days for albumin) and a smaller body pool than albumin, making the former more rapidly responsive than albumin.10 Additionally, due to its high percentage of tryptophan and essential amino acids, prealbumin is more sensitive to protein nutrition than albumin and is less affected by liver disease or hydration state than albumin.11 In practice, prealbumin is generally used to assess protein calorie nutrition. Prealbumin is generally regarded as the best laboratory test of protein nutrition, and it is routinely used to monitor patients receiving IV or tube feeding.12–14
International Normalized Ratio and Prothrombin Time
Normal range: INR 0.9–1.1, PT 12.7–15.4 sec
For an introduction to international normalized ratio (INR) and prothrombin time (PT), please see Chapter 16: Hematology: Blood Coagulation Tests. These two tests measure the speed of a set of reactions in the extrinsic pathway of the coagulation cascade. A coagulation deficit correlates with prolonged reaction times, and increased values of INR and PT. Both PT and INR are two different measures of the same set of reactions, with the INR being a derived index that takes into account variations between test reagents used in different laboratories. As such, INR is more precise and easily interpretable, and it is replacing the use of the PT.
The liver is required for the synthesis of clotting factors (with the exception of factor VIII), many of which require a vitamin K cofactor for their activation. Therefore, either hepatic impairment or vitamin K deficiency may lead to a deficiency in activated coagulation factors with subsequent prolongation of PT/INR. Both synthetic failure and vitamin K deficiency may also cause prolongation of activated partial thromboplastin time (aPTT which measures a different set of coagulation reactions in the intrinsic coagulation cascade) but to a much lesser degree than PT/INR.
The prolongation of PT/INR alone is not specific for liver disease. It can be seen in many situations, most of which interfere with the utilization of vitamin K, a cofactor required for the proper posttranslational activation of clotting factors II, VII, IX, and X. Because vitamin K is a fat soluble vitamin, inadequate vitamin K in the diet or fat malabsorption as caused by cholestasis may cause hypovitaminosis.15 Many broad-spectrum antibiotics, including tetracyclines, may eliminate vitamin K-producing flora in the gut. The anticoagulant agent warfarin interferes directly with vitamin K dependent activation of clotting factors.
If the etiology of elevated PT/INR remains unclear despite obtaining additional coagulation tests, then the clinical approach is to provide parenteral vitamin K (generally subcutaneously).16 If the PT/INR is prolonged due to malabsorption, warfarin, perturbed gut flora, or the absence of vitamin K in the diet, the PT/INR usually corrects by at least 30% within 24 hours.9 Alternatively failure of PT/INR to normalize despite parenteral vitamin K suggests impaired synthetic liver function (see Figure 12-2).4 Other factors that may cause a prolonged PT/INR that does not respond to parenteral vitamin K include inherited clotting factor deficiencies.
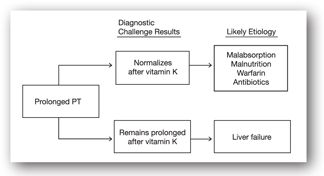
FIGURE 12-2. Evaluation of a prolonged PT.
Because clotting factors are produced in excess of need and because the liver has tremendous synthetic reserves, only substantial hepatic impairment (>80% loss of synthetic capability) leads to decreased synthesis of these factors and subsequent clotting abnormalities.6 Thus, PT/INR, albumin, and prealbumin levels lack sensitivity and may remain normal in the face of substantial liver damage.4 However, they have considerable prognostic value if liver damage is sufficient to affect them. Unlike albumin (which responds slowly to hepatic insult), PT responds within 24 hours to changes in hepatic status due to the short half-life of certain clotting proteins (i.e., factor VII has a half-life of less than 6 hours).17 Thus, the PT may become elevated days before other manifestations of liver failure and, likewise, may normalize prior to other evidence of clinical improvement.18 The primary utility of PT in liver disease is to provide prognostic data, generally in situations where the cause of the elevated PT/INR is known, for example, acute acetaminophen overdose leading to hepatic failure.
In addition to serving as a liver function test, PT/INR has direct clinical relevance in accessing the patient’s tendency to bleed spontaneously, or due to surgical or diagnostic procedures. Bleeding is a dramatic complication of hepatic failure. When the PT/INR is significantly elevated, bleeding may be controlled or at least diminished by fresh frozen plasma, which contains the needed activated clotting factors and often corrects the PT/INR temporarily.
Des-gamma-carboxy prothrombin (DCP) is an abnormal prothrombin form that is released in the absence of vitamin K, in the presence of vitamin K antagonists (warfarin), and by certain tumors (hepatocellular carcinoma). White it has been used to evaluate the risk of hepatocellular carcinoma, DCP has a lower sensitivity than alpha fetoprotein for tumors under 3 cm. in size.19
CHOLESTATIC LIVER DISEASE
Cholestasis is a deficiency of the excretory function of the liver. As described above, bile is normally secreted by hepatocytes into bile canaliculi, where it flows into larger bile ducts, and eventually empties into the duodenum. Excretion of bile from the liver serves multiple purposes. Certain large lipophilic toxins, drugs, and endogenous substances are eliminated by secretion into the bile with eventual elimination in the feces. Bile salts also play an important role in dissolving and absorbing dietary fat-soluble vitamins and nutrients within the small intestine.
Failure of the excretory functions of the liver leads to a predictable set of consequences. Substances normally secreted in the bile accumulate, resulting in jaundice (from bilirubin), pruritus (from bile salts), or xanthomas (from lipid deposits in skin). Absence of bile salts to dissolve fat-soluble nutrients leads to deficiencies of fat-soluble vitamins A, D, E, and K. This may result in osteoporosis, due to lack of vitamin D, and PT/INR elevation, due to lack of vitamin K.
Cholestatic syndromes may be subclassified as either anatomic obstructions to macroscopic bile ducts (extrahepatic cholestasis) or disorders of hepatocytes and microscopic bile ducts (intrahepatic cholestasis).20 The approach to a patient with cholestasis generally begins with a radiographic study, often a right upper-quadrant ultrasound, to look for dilation of large bile ducts within or outside the liver. Dilation of such large bile ducts indicates extrahepatic cholestasis; otherwise extrahepatic cholestasis is largely excluded, and the next step is to investigate for various causes of intrahepatic cholestasis.
Intrahepatic Cholestasis
Intrahepatic cholestasis includes a variety of processes that interfere with hepatocyte secretion of bile, as well as diseases of the micro- and macroscopic bile ducts within the liver. Etiologies involving impaired hepatocyte secretion of bile overlap to some extent with hepatocellular diseases as noted above; such processes include viral hepatitis (especially type A), alcoholic hepatitis, and even cirrhosis. Processes that cause a more pure cholestatic pattern include a variety of drugs (see Table 12-2), pregnancy, severe infection (cholestasis of sepsis), and certain nonhepatic neoplasms, especially renal cell carcinoma. Infiltrative process of the liver will produce a primarily cholestatic pattern, and these include granulomatous diseases and amyloidosis. Primary biliary cirrhosis causes inflammatory scarring of the microscopic bile ducts, whereas sclerosing cholangitis is a similar process that may affect micro- or macroscopic bile ducts within the liver. Masses within the liver, including tumors or abscesses, may block the flow of bile as well.
ACE = angiotensin-converting enzyme; AIDS = acquired immune deficiency syndrome; INH = isoniazid; NAFLD = nonalcoholic fatty liver disease; NSAIDs = nonsteroidal anti-inflammatory drugs; PBC = primary biliary cirrhosis; PSC = primary sclerosing cholangitis; TB = tuberculosis; TPN = total parenteral nutrition.
aPlease note that listings of drugs contain more commonly used agents and are not exhaustive. For any particular patient, potentially causative drugs should be specifically researched in appropriate databases to determine any hepatotoxic effects.
Extrahepatic cholestasis involves obstruction of the large bile ducts outside of the liver. The most common cause is stones in the common bile duct; other causes include obstruction by strictures (after surgery), or tumors (of pancreas, ampulla of Vater, duodenum, or bile ducts), chronic pancreatitis with scarring of the ducts as they pass through the pancreas, and parasitic infections of the ducts. Another cause is sclerosing cholangitis, a disease causing diffuse inflammation of the bile ducts, often both intrahepatic and extrahepatic. Of note is that sclerosing cholangitis is associated with inflammatory bowel disease, especially involving the colon. Some patients with HIV can develop a picture similar to sclerosing cholangitis, referred to as AIDS cholangiopathy. Although previously referred to as surgical cholestasis, extrahepatic cholestasis can now often be treated or at least palliated using endoscopic means (e.g., dilation of strictures with or without stent placement).
Tests Associated with Excretory Liver Function and Cholestasis
Laboratory tests do not distinguish between intra- and extrahepatic cholestasis. This distinction is usually made radiographically. In most instances of extrahepatic cholestasis, a damming effect causes dilation of bile ducts above the obstruction, which can be visualized via a computed tomography (CT) scan, a magnetic resonance imaging (MRI) scan, or ultrasound. Laboratory abnormalities primarily associated with cholestasis include elevation of ALP, 5’-nucleotidase, gamma-glutamyl transpeptidase (GGT), and bilirubin.
Alkaline Phosphatase
Normal range3: 33–96 units/L
Alkaline phosphatase (ALP) refers to a group of isoenzymes whose exact function remains unknown. These enzymes are found in many body tissues including the liver, bone, small intestine, kidneys, placenta, and leukocytes. In the liver they are found primarily in the bile canicular membranes of the liver cells. In adults, most serum ALP comes from the liver and bone (~80%), with the remainder mostly contributed by the small intestine.
Normal ALP concentrations vary primarily with age. In children and adolescents, elevated ALP concentrations result from bone growth, which may be associated with elevations as high as 3 times the adult normal range. Similarly, the increase during late pregnancy is due to placental ALP.21,22 In the third trimester, concentrations often double and may remain elevated for 3 weeks postpartum.23
The mechanism of hepatic ALP release into the circulation in patients with cholestatic disease remains unclear. Bile accumulation appears to increase hepatocyte synthesis of ALP, which eventually leaks into the bloodstream.8,24 The ALP concentrations persist until the obstruction is removed and then normalize within 2–4 weeks.
Clinically, ALP elevation is associated with cholestatic disorders and, as mentioned previously, does not help to distinguish between intra- and extrahepatic disorders. ALP concentrations more than 4 times normal suggest a cholestatic disorder, and 75% of patients with primarily cholestatic disorders have ALP concentrations in this range (Table 12-3). Concentrations of 3 times normal or less are nonspecific and can occur in all types of liver disease. Mild elevations, usually less than 1½ times normal, can be seen in normal patients and are less significant.
| TABLE 12-3. Initial Evaluation of Elevated ALP Concentrations in Context of Other Test Results | |||
| ALP | GGT, 5’ NUCLEOTIDASE | AMINOTRANSFERASES (ALT AND AST) | DIFFERENTIAL DIAGNOSIS |
| Mildly elevated | Within normal limits | Within normal limits | Pregnancy; nonhepatic causes (Table 12-4) |
| Moderately elevateda | Markedly elevated | Within normal limits or minimally elevated | Cholestatic syndromes |
| Mildly elevatedb | Mildly elevated | Markedly elevated | Hepatocellular disease |
aUsually greater than 4 times normal limits.
bUsually less than 4 times normal limits.
When faced with an elevated ALP concentration, a clinician must determine whether it is derived from the liver. One approach is to fractionate the ALP isoenzymes using electrophoresis, but this method is expensive and often unavailable. Thus, the approach usually taken is to measure other indicators of cholestatic disease, 5’-nucleotidase or GGT. If ALP is elevated, an elevated 5’-nucleotidase or GGT indicates that at least part of the elevated ALP is of hepatic origin. Alternatively, a normal 5’-nucleotidase or GGT suggests a nonhepatic cause (Table 12-3).
Nonhepatic causes of elevated ALP include bone disorders (e.g., healing fractures, osteomalacia, Paget disease, rickets, tumors, hypervitaminosis D, or vitamin D deficiency as caused by celiac sprue), hyperthyroidism, hyperparathyroidism, sepsis, diabetes mellitus, renal failure, and neoplasms (which may synthesize ALP ectopically, outside tissues that normally contain ALP) (Table 12-4). Some families have inherited elevated concentrations (2–4 times normal), usually as an autosomal dominant trait.25 Markedly elevated concentrations (greater than 4 times normal) are generally seen only in cholestasis, Paget disease, or infiltrative diseases of the liver. Due to an increase in intestinal ALP, serum ALP concentrations can be falsely elevated in patients of blood type O or B whose blood is drawn 2–4 hours after a fatty meal.26
Alkaline phosphatase concentrations can be lowered by a number of conditions including hypothyroidism, hypophosphatemia, pernicious anemia, and zinc or magnesium deficiency.8 Also, ALP may be confounded by a variety of drugs.
5’-Nucleotidase
Normal range: 0–11 units/L
Although 5’-nucleotidase is found in many tissues (including liver, brain, heart, and blood vessels), serum 5’-nucleotidase is elevated most often in patients with hepatic diseases.19 It has a response profile parallel to ALP and similar utility in differentiating between hepatocellular and cholestatic liver disease. Since it is only elevated in the face of liver disease, the presence of an elevated ALP in the face of a normal 5’-nucleotidase suggests that the ALP is elevated secondary to nonhepatic causes.
Gamma-Glutamyl Transpeptidase
Normal range: 9–58 units/L
Gamma-glutamyl transpeptidase (GGT, also GGTP), a biliary excretory enzyme, can also help determine whether an elevated ALP is of hepatic etiology. Similar to 5’-nucleotidase, it is not elevated in bone disorders, adolescence, or pregnancy. It is rarely elevated in conditions other than liver disease.
Generally, GGT parallels ALP and 5’-nucleotidase levels in liver disease. Additionally, GGT concentrations are usually elevated in patients who abuse alcohol or have alcoholic liver disease. Therefore, this test is potentially useful in differential diagnosis with a GGT/ALP ratio greater than 2.5 being highly indicative of alcohol abuse.16,27 With abstinence, GGT concentrations often decrease by 50% within 2 weeks.
Although GGT is often regarded as the most sensitive test for cholestatic disorders, unlike 5’-nucleotidase it lacks specificity. In one study of nonselected patients, only 32% of GGT elevations were of hepatic origin.18 Gamma-glutamyl transpeptidase is found in the liver, kidneys, pancreas, spleen, heart, brain, and seminal vesicles. Elevations may occur in pancreatic diseases, myocardial infarction, severe chronic obstructive pulmonary diseases, some renal diseases, systemic lupus erythematosus, hyperthyroidism, certain cancers, rheumatoid arthritis, and diabetes mellitus. Gamma-glutamyl transpeptidase may be confounded in patients on a variety of medications, some of which overlap with the medications that confound ALP test results. Thus, elevated GGT (even with concomitant elevated ALP) does not necessarily imply liver injury when 5’-nucleotidase is normal, but rather both elevations in GGT and ALP may be due to a common confounding medication (e.g., phenytoin, barbiturates) or medical conditions (e.g., myocardial infarction).
Bilirubin
Total bilirubin: 0.3–1.3 mg/dL
Indirect (unconjugated, insoluble) bilirubin: 0.2–0.9 mg/dL (bound to albumin in the blood)
Direct (conjugated, water soluble) bilirubin3: 0.1–0.4 mg/dL (excreted by the kidney)
Understanding the various laboratory studies of bilirubin requires knowledge of the biochemical pathways for bilirubin production and excretion (see Figure 12-3). Bilirubin is a breakdown product of heme pigments, which are large, insoluble organic compounds. Most of the body’s heme pigments are located in erythrocytes, where they are a component of hemoglobin. Breakdown of erythrocytes releases hemoglobin into the circulation, which is converted to bilirubin (predominantly in the spleen), where it is initially a large lipophilic molecule bound to albumin.
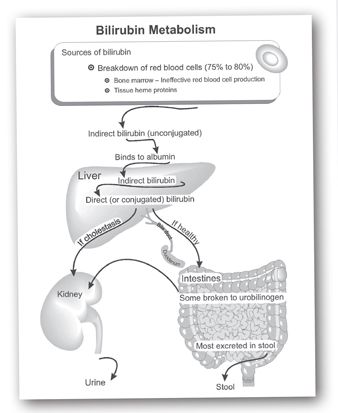
FIGURE 12-3. Overview of bilirubin production and metabolism. Most bilirubin is produced by the breakdown of heme pigments in erythrocytes (red blood cells) and to a lesser extent other tissues. The indirect bilirubin is carried in the circulation to the liver where it is conjugated and becomes direct or conjugated bilirubin. In health this is largely excreted via the biliary system into the gut. In disease it will “back up” into the circulation, causing elevated levels of direct/conjugated bilirubin and ultimately jaundice. (With thanks to Esta Farkas.)
The liver plays a central role in excretion of bilirubin, similar to its role in the metabolism and excretion of a wide variety of lipophilic substances. Prior to excretion, bilirubin must be converted into a form that is water-soluble. The liver achieves this covalently linking it to a water-soluble sugar molecule (glucuronic acid) using an enzyme glucuronyl transferase. The conjugate of bilirubin linked to glucuronic acid is water soluble, so it may then be excreted into the bile and eventually eliminated in the feces. Incidentally, bilirubin and some of its breakdown products are responsible for coloring feces brown (such that with complete obstruction of the bile ducts or cessation of bile synthesis by the liver, stool will take on a pale color).
Indirect Versus Direct Bilirubin
The total amount of bilirubin in the serum can be divided into direct and indirect fractions. Bilirubin conjugated to glucuronic acid (water-soluble bilirubin) reacts quickly in the van der Bergh reaction and is thus called direct-reacting or direct bilirubin. Alternatively, unconjugated bilirubin, because it is water insoluble, requires the presence of dissolving agents to be detected by this assay and is thus called indirect-reacting or indirect bilirubin. Although this nomenclature system is slightly awkward, it is the standard terminology used in clinical practice today. Only the water-soluble direct bilirubin can be excreted in the urine, and therefore urine dipsticks will only measure this fraction. In fact, urine dipsticks may be more sensitive than most serum tests for detecting a slight elevation of direct bilirubin.
Elevated bilirubin causes abnormal yellow coloration of the skin ( jaundice) and sclera of the eyes (icterus). Excess carotenes (as due to large amounts of carrot consumption) may cause a similar effect on the skin but spare the eyes. Icterus usually becomes visible when total bilirubin concentrations exceed 2–4 mg/dL. In infants, extremely elevated concentrations of bilirubin (for example, >20 mg/dL) may have neurotoxic effects on the developing brain, but in adults a direct toxic effect of bilirubin is quite rare.28
The first step in evaluating an elevated serum bilirubin is to determine if only the indirect fraction is elevated, or if there is involvement of the direct fraction. Given the sequential location of these two molecules within the pathway of bilirubin metabolism, elevated levels of the molecules may have markedly different significance (Table 12-5).
ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma-glutamyl transpeptidase.
aUsually indirect bilirubin is less than 4 mg/dL but may increase to 18 mg/dL.
bUsually indirect bilirubin is greater than 12 mg/dL and may go as high as 45 mg/dL.
cThese syndromes are distinguished in the laboratory by liver biopsy.
dUsually direct bilirubin is 3–10 mg/dL.
Indirect Hyperbilirubinemia (unconjugated, insoluble)
Indirect bilirubin is mostly produced by the breakdown of erythrocytes and is removed from the circulation by conversion to direct bilirubin by glucuronyl transferase in the liver. Therefore, elevated levels may result from increased breakdown of red blood cells (hemolysis) or reduced hepatic conversion to direct bilirubin.18 Patients with primarily unconjugated hyperbilirubinemia (>70% indirect) generally do not have serious liver disease. The most common causes of elevated indirect bilirubin are hemolysis, Gilbert syndrome, Crigler-Najjar syndrome, or various drugs, including probenecid and rifampin.29 In infants this can be physiologic (neonatal jaundice), although very high levels may require medical intervention.
Hemolysis refers to increased destruction of erythrocytes, which increases the production of indirect bilirubin and may overwhelm the liver’s ability for conjugation and excretion. However, the liver’s processing mechanisms are intact so that serum bilirubin generally doesn’t climb too high (rarely >5 mg/dL). Hemolysis may result from a wide variety of hematologic processes including sickle cell anemia, spherocytosis, hematomas, mismatched blood transfusions, or intravascular fragmentation of blood cells. Evaluation will include various hematologic tests as described in further detail in another chapter.
Gilbert syndrome is an inherited, benign trait present in 3% to 5% of the population. It is due to reduced production of hepatic glucuronyl transferase enzymes, resulting in intermittent elevation of indirect bilirubin and mild jaundice (increased with fasting, stress, or illness). The primary significance is that it may cause elevation of bilirubin when there is in fact no significant hepatic or hematologic disease. Bilirubin elevation is generally mild, with values less than 5 mg/dL.2
Direct Hyperbilirubinemia (conjugated, soluble)
Conjugated hyperbilirubinemia is defined as bilirubinemia with >50% in the direct fraction (although absolute levels of unconjugated bilirubin may also be elevated).17 In the normal course of bilirubin metabolism, direct bilirubin is synthesized in hepatocytes and secreted into bile. Therefore, elevated direct bilirubin implies hepatic or biliary tract disease that interferes with secretion of bilirubin from the hepatocytes or clearance of bile from the liver.
Direct hyperbilirubinemia is generally classified as a positive cholestatic liver test, although as discussed earlier, it may be elevated to some extent in hepatocellular processes as well. In cholestatic disease bilirubin is primarily conjugated, whereas in hepatocellular processes significant increases in both conjugated and unconjugated bilirubin may result. The most reliable method of determining the cause of hyperbilirubinemia considers the magnitude and pattern of abnormalities in the entire liver function panel. It should be noted that direct bilirubin is generally readily cleared by the kidney, such that its levels never rise very high even in severe cholestatic disease if the patient has normal renal function. Very rarely, congenital disorders (e.g., Dubin-Johnson and Rotor syndromes) may cause elevations of primarily conjugated bilirubin.
It should be noted that a gray area exists between indirect and direct hyperbilirubinemia. Most authors agree that greater than 50% direct bilirubin indicates direct hyperbilirubinemia whereas less than 30% direct fraction indicates indirect hyperbilirubinemia.2 For cases where the fraction falls between 30% to 50%, other liver tests and hematologic tests may be required to determine the etiology. Patients with elevated direct bilirubin levels may have some binding of the bilirubin to the albumin, referred to as delta bilirubin. This explains delayed resolution of jaundice during recovery from acute hepatobiliary diseases, for while the “free” bilirubin is rapidly metabolized, the bilirubin linked to albumin is metabolized at a much slower rate. Delta bilirubin has a half-life similar to albumin, of 14–21 days.30,
HEPATOCELLULAR INJURY
As discussed earlier, the liver is a large organ with diverse biochemical roles, which require its cells to be in close communication with the bloodstream. These properties place the hepatocytes at risk of injury due to a variety of processes. Toxin and drug metabolism produce cascades of metabolic byproducts, some of which may damage hepatocytes. Likewise, the liver plays a central role in the body’s biochemical homeostasis, so metabolic disorders tend to involve the liver. Finally, the close relationship of the hepatocytes to the blood supply places them at risk for a variety of infectious agents.
Hepatitis is a term that technically refers to a histologic pattern of inflammation of hepatocytes. It may also be used to refer to a clinical syndrome due to diffuse liver inflammation. The laboratory reflection of hepatitis is a hepatocellular injury pattern, which is marked primarily by elevated aminotransferases.
There are multiple causes of hepatitis. One common type is viral hepatitis, which is classified A, B, C, D (delta hepatitis), E, or G based on the causative virus. These viruses, and the tests for them, are discussed in detail in the Viral Hepatitis section. Less commonly viral hepatitis may be caused by the Epstein-Barr virus, herpes virus, or cytomegalovirus.
Hepatitis may also be caused by various medications, and such drug-induced hepatitis can be either acute or chronic.28 Some drugs commonly implicated in cellular hepatotoxicity are listed in Table 12-2. In addition, elevation of aminotransferases has been reported in patients receiving heparin.31 ALT is elevated in up to 60% of these patients, with a mean maximal value of 3.6 times the baseline. A vast number of drugs can cause hepatic injury, especially drugs that are extensively metabolized by the liver. Although numerous drugs may result in aminotransferase elevations, such elevations are usually minor, transient, not associated with any symptoms, and of no clinical consequence.32
Perhaps the most common cause of abnormal aminotransferases in ambulatory patients is that of fatty liver. Estimates are that 30% to 40% of adults in the United States have fatty liver, which can vary from hepatic steatosis (fat in the liver) to nonalcoholic steatohepatitis (NASH), where the extra fat in the liver is associated with inflammation. It is potentially serious as up to one-quarter of these patients can progress to cirrhosis. Fatty liver and NASH are mostly related to increased body mass index (BMI), but they can also be associated with rapid weight loss or drugs such as tamoxifen, amiodarone, diltiazem, nifedipine, corticosteroids, and petrochemicals. Fatty liver/NASH can also be seen in hepatitis C and patients on total parenteral nutrition (TPN), and it is associated with hypothyroidism and short bowel syndrome. It is important to note that although mild hepatic inflammation is often of minimal significance, it may signal the presence of a chronic and serious disease process. Some other causes of hepatic inflammation and injury are listed in Table 12-2.
It is often difficult to determine the exact etiology of hepatic inflammation or hepatitis. A careful history—especially for exposure to drugs, alcohol, or toxins—and detailed physical examination are crucial. Additional laboratory studies are usually necessary to distinguish one form of hepatitis from another (Figure 12-4). Radiological testing or liver biopsy may be indicated, not only to determine the etiology of the liver disease, but also to help determine the indications for (and results of) therapy and prognosis.
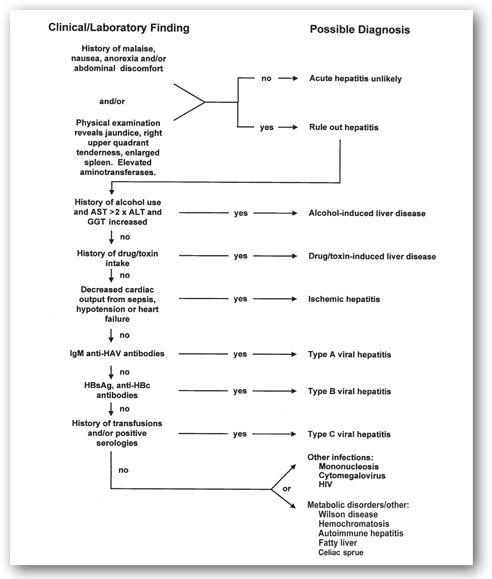
FIGURE 12-4. Algorithm for differential diagnosis of suspected hepatitis.
Aminotransferases: AST and ALT
AST: 12–38 units/L; ALT: 7–41 units/L (normal values vary from lab to lab but tend to be in the range of <30 units/L for men and <20 units/L for women)
The aminotransferases (also known as transaminases) are used to assess hepatocellular injury. These enzymes are primarily located inside hepatocytes, where they assist with various metabolic pathways. They are released into the serum in greater quantities when there is hepatocyte damage, and they are very sensitive and may be elevated even with minor levels of hepatocyte damage.33 However, this renders them relatively nonspecific, and slightly elevated levels may not be clinically significant (particularly in an ill, hospitalized patient who is on many medications and has a variety of active medical problems).
Aminotransferases will often be slightly increased in cholestatic liver diseases, but in this situation they will generally be overshadowed by a greater elevation of cholestatic liver tests (i.e., ALP and total bilirubin to produce a predominantly cholestatic pattern of liver tests). If both transaminases and cholestatic tests are elevated in a similar pattern, it suggests a severe hepatocellular process, which interferes with bile secretion at the level of the hepatocytes. Finally, it should be noted that aminotransferases may rise into the thousands within 24–48 hours following common bile duct obstruction, after which they decline rapidly. This is one instance in which a cholestatic process may transiently cause a hepatocellular injury LFT profile.
Both AST and ALT have half-lives of 17 and 47 hours, respectively, so they reflect active hepatocyte damage, and not, for example, damage to hepatocytes that occurred weeks, months, or years previously. This may lead to some counterintuitive relationships between aminotransferase levels and the overall state of the liver. For example, a drop in aminotransferase levels in the setting of acute massive (fulminant) hepatitis may reflect a depletion of viable hepatocytes with poor prognosis.18 Extremely high concentrations (>1000 International Units/L) are usually associated with acute viral hepatitis, severe drug or toxic reactions, or ischemic hepatitis (inadequate blood flow to the liver). Lesser elevations are caused by a vast number of hepatic insults and are less specific.34
The ratio of AST to ALT may be of value in diagnosing alcoholic hepatitis, where the AST is generally at least twice the ALT and the AST is rarely above 300 International Units/L. In alcoholic liver disease, a mitochondrial isoform of AST with a relatively long half-life (87 hours) is released from hepatocytes, increasing the AST/ALT ratio. Alcoholic liver disease is also suggested by an elevation in GGT as previously reviewed.
Aspartate aminotransferase is not solely located in hepatocytes but rather is also found in cardiac muscle, skeletal muscle, kidneys, brain, lungs intestines, and erythrocytes. Consequently, AST may be elevated due to a variety of situations including musculoskeletal diseases (e.g., muscular dystrophy, dermatomyositis, heavy exercise, trichinosis, gangrene, and muscle damage secondary to hypothyroidism), myocardial infarction, renal infarction or failure, brain trauma or cerebral infarction, hemolysis, pulmonary embolism, necrotic tumors, burns, and celiac sprue.6,7,18 Alanine aminotransferase is more localized to the liver than AST, so it is more specific to liver injury. Elevation of AST without elevation of the ALT or other liver test abnormality suggests cardiac or muscle disease.17 A muscular origin of aminotransferases may also be indicated by increases in aminotransferases above 300 International Units/L with concomitant increases in serum creatine kinase (CK) activity.25,35
Measurement of AST may be affected by a bewildering variety of medications. Almost any prescription drug (as well as various herbal compounds and illegal drugs) can cause an elevation of aminotransferases, and the significance of these elevations is often unclear.26 Furthermore, the in vitro assay may be confounded by a variety of factors including uremia, hyperlipidemia, and hemolysis.36 False elevations in the in vitro test may be also seen in patients on acetaminophen, levodopa, methyldopa, tolbutamide, para-aminosalicylic acid, or erythromycin.8,37,38
Other factors may interfere with the test’s accuracy. Levels may be elevated to 2–3 times normal by vigorous exercise in males and decreased to about half following dialysis.7 Complexing of AST with immunoglobulin (known as macro-AST) may occasionally produce a clinically irrelevant elevation of AST. Testing for macro-AST is not a clinical lab test used in practice. Given the array of factors that can cause an abnormal result, unexplained false positives often occur. In healthy individuals, an isolated elevated ALT returns to normal in repeat studies one-half to one-third of the time.39 For this reason, prior to an evaluation of mildly elevated aminotransferases in low-risk healthy patients, a practitioner should see either elevation of more than one test (i.e., both AST and ALT) or repeated elevations of a single test.
TESTS ASSOCIATED WITH DETOXIFICATION
Hepatic Encephalopathy
Hepatic encephalopathy refers to a diffuse metabolic dysfunction of the brain that may occur in acute or chronic liver failure. Clinically it ranges from subtle changes in personality to coma and death.
The etiology of hepatic encephalopathy remains controversial and has undergone significant revision recently. Many theories ascribe a major role to ammonia. The majority of serum ammonia enters the blood from the intestines, where it is formed by bacterial catabolism of protein within the gut lumen as well as conversion of serum glutamine into ammonia by enterocytes of the small intestine.40 Normally, the liver removes >90% of this ammonia via first-pass metabolism.41 In liver failure, ammonia, along with possibly other toxic substances may avoid this first-pass metabolism and gain immediate access to the brain where it has a variety of toxic effects.42 While serum ammonia is currently the “standard” lab test for assessing hepatic encephalopathy, other tests are being developed. Elevated serum levels of 3-nitro-tyrosine may be a marker for minimal hepatic encephalopathy (cirrhotic patients with mild cognitive impairment). In a pilot study using a cutoff of 14 nm, 3-nitro-tyrosine levels had a 93% sensitivity and an 89% specificity in identifying these patients.43
Ammonia
Normal range3: 19–60 mcg/dL
Ammonia levels do not correlate well with hepatic encephalopathy in the setting of chronic liver failure (i.e., patients with cirrhosis). This is likely because hepatic encephalopathy also involves an increase in the permeability of the blood–brain barrier to ammonia.6,44 There is a large overlap between ammonia levels in patients with and without hepatic encephalopathy among patients with chronic liver disease making this a poor test in this situation.45 A very high ammonia level (i.e., greater than 250 mcg/dL) is suggestive of hepatic encephalopathy, but most cirrhotic patients suspected of having encephalopathy will have normal or slightly elevated ammonia levels, which adds little diagnostic information. In this situation, hepatic encephalopathy is a clinical diagnosis based on history, physical exam, and exclusion of other possibilities.
Some recent studies have suggested that ammonia may have more significance in the setting of acute liver failure (e.g., due to overwhelming infection of the liver by viral hepatitis). In these patients, the degree of ammonia elevation correlates with severity of hepatic encephalopathy and the likelihood of death, and may be a useful marker for predicting which patients require emergent liver transplant.46,47
Ammonia concentration may also be elevated in patients with Reye syndrome, inborn disorders of the urea cycle, various medications (most notably valproic acid), impaired renal function, uterosigmoidostomy, or urinary tract infections with bacteria that convert urea to ammonia. In cirrhotic patients or patients with mild liver disease, elevated ammonia and hepatic encephalopathy may be precipitated by such factors as increased dietary protein, GI bleeding, constipation, and Helicobacter pylori infection.7
VIRAL HEPATITIS
The onset of acute viral hepatitis may be quite dramatic and present as an overwhelming infection, or it may pass unnoticed by the patient. In the usual prodromal period, the patient often has a nonspecific flu-like illness that may include nausea, vomiting, fatigue, or malaise. This period may be followed by clinical hepatitis with jaundice. During this time, the most abnormal laboratory studies are usually the aminotransferases, which can be in the thousands (normal values vary, in range of <30 units/L for men and <20 units/L for women). Bilirubin may be quite elevated, while ALP only mildly so.
The major types of viral hepatitis are reviewed here, but they are often clinically indistinguishable. Thus, serologic studies of antibodies, molecular assays to detect viral genetic material, and knowledge of the epidemiology and risk factors for different viruses (Table 12-6) are central to diagnosis.
HBV = hepatitis B virus; HCV = hepatitis c virus; IV = intravenous.
Type A Hepatitis
Hepatitis A virus (HAV) is spread primarily by the fecal-oral route by contaminated food or water or by person-to-person contact. It has an incubation period of 3–5 weeks with a several-day prodrome (preicteric phase) before the onset of jaundice and malaise, or the icteric phase. The icteric phase generally lasts 1–3 weeks, although prolonged courses do occur. Hepatitis A is responsible for about 50% of acute hepatitis in the United States (more than all other hepatotropic viruses combined), generally due to person-to-person contact within community-wide outbreaks.48,49 Interestingly the incidence of new cases has diminished with the wide spread use of the HAV vaccine (see below).
Unlike types B, C, and D hepatitis virus, HAV does not cause chronic disease, and recovery usually occurs within one month. Many patients who get type A hepatitis never become clinically ill. Perhaps 10% of all patients become symptomatic, and only 10% of those patients become jaundiced.4 The majority of patients have a full recovery, but there is a substantial mortality risk in elderly patients, very young patients, as well as in patients with chronic hepatitis B or C and patients with chronic liver disease of other etiologies.50-52
A vaccine for HAV is now available. It is recommended for those traveling to endemic regions (i.e., Central and South America), men who have sex with men, users of street drugs, those with occupational exposure, susceptible patients requiring clotting factors, those from endemic regions, and patients with chronic liver diseases. This vaccine is increasingly being recommended as a universal vaccine for pediatric patients. While this vaccine is generally preferred for postexposure prophylaxis, use of immunoglobulin should be considered in the very young (under 12 months) or patients who cannot use the vaccine.
Presently, the only two tests available measure antibodies to HAV, either immunoglobulin M (IgM) or total (all isotypes of) antibody. Detection of IgM is the more clinically relevant test as it reveals acute or recent infection. These antibodies are present at the onset of jaundice and decline within 12 (usually 6) months.52 Total antibody, which is comprised of antibody of all isotypes against HAV, indicates present or previous infection or immunization (Figure 12-5).
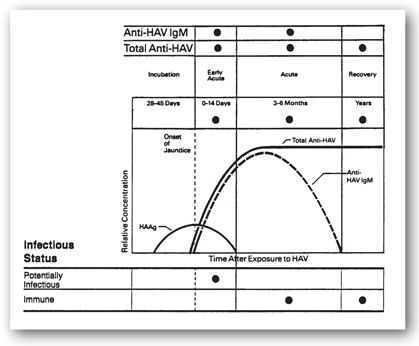
FIGURE 12-5. Temporal relationships of serologies for type A hepatitis with onset of jaundice and infectious status. Anti-HAV IgM is the IgM antibody against the hepatitis A virus. HAAg is the hepatitis A antigen (virus). Total anti-HAV is primarily IgG antibodies (and some IgM in acute phase) against hepatitis A virus. (Adapted with permission from educational material of Abbott Laboratories, North Chicago, IL.)
Type B Hepatitis
Hepatitis B virus (HBV) is a DNA virus spread by bodily fluids, most commonly as a sexually transmitted disease, but also via contaminated needles (as with drug abuse or needle stick accidents), shared razor blades or toothbrushes, nonsterile tattooing or body piercing, blood products, or vertical transmission (transmission from mother to child, generally at birth). This disease is 50–100 times more contagious than HIV. The incubation period of HBV varies from 2–4 months, much longer than that of HAV. Geographically, there is a markedly increased prevalence of hepatitis B in Southeast Asia, China, and sub-Saharan Africa with 10% to 20% of the populations being hepatitis B carriers. In contrast, the incidence of hepatitis B carriers in the United States is approximately 0.5%.
The clinical illness is generally mild and self-limited but can be quite severe. Unfortunately, up to 5% of infected adults and 90% of infected neonates develop a chronic illness. Chronic HBV infection is often mild but may progress to cirrhosis, liver failure, or hepatocellular carcinoma, thereby contributing to premature death in 15% to 25% of cases.53
Viral Antigens and Their Antibodies
Three HBV antigens and antibody systems are relevant to diagnosis and management (HBsAg, HBcAg, and HBeAg). The HBV surface antigen (HBsAg) is present on the outer surface of the virus, and neutralizing antibodies directed against this protein (anti-HBs) are central to natural and vaccine-induced immunity (Figure 12-6).
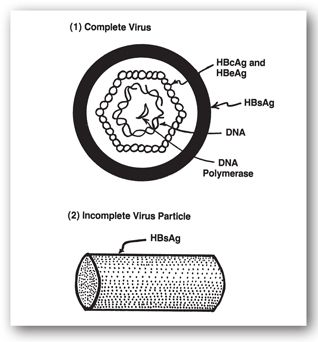
FIGURE 12-6. Hepatitis B virus and its antigenic components. The complete and infectious virus (1), originally known as the Dane particle, is composed of the outer layer (HBsAg) and inner nucleocapsid core. The inner core is comprised of HBcAg intermeshed with HBeAg and encapsulates the viral DNA. HBeAg may be an internal component or degradation product of the nucleocapsid core. An incomplete and noninfectious form (2) is composed exclusively of HBsAg and is cylindrical in shape.
Neither the core protein (HBcAg) nor the e antigen (HBeAg) are on the surface of the virion, and thus antibodies against these antigens are not protective. Nevertheless, antibodies are directed against these proteins and may serve as markers of infection. Of these antigens, only HBsAg and HBeAg can be detected in the serum by conventional techniques.53 HBsAg is detected for a greater window of time during infection and reveals active infection. Detection of HBeAg indicates large amounts of circulating hepatitis B virus; these patients are 5–10 times more likely to transmit the virus than are HBeAg negative persons. HBsAg levels are often used to determine a given patient’s suitability for hepatitis B therapy, and subsequently to monitor for effectiveness of hepatitis B therapy.
In response to infection with HBV, the body may produce antibodies to the antigens: antisurface antibody (anti-HBs), anticore antibody (anti-HBc), and anti-e antibody (anti-HBe). All of these antibodies can be detected in clinical laboratories, and in the case of anti-HBcAg separate tests are available to detect IgM or total antibody (all isotypes). Anti-HBs is associated with resolved type B hepatitis, or patients who have responded to vaccination for HBV. Anti-HBc is a bit more challenging to interpret, as it can be seen in acute type B hepatitis, after recovery from type B hepatitis (often in concert with Anti-HBs), in chronic infection (often with HBsAg, and HBeAg), and there can be false-positive results as well. As shown in Table 12-7 and Figure 12-7, levels of antigens and antibodies show complex patterns in the course of HBV infection and thus can yield considerable information about the infection’s course and chronology (Table 12-7).
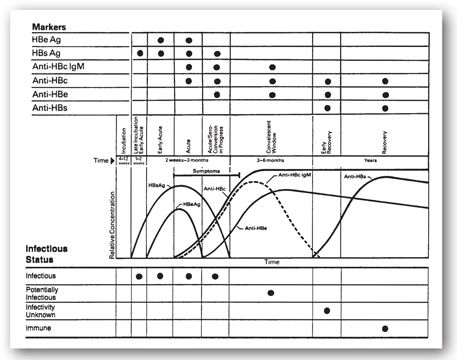
FIGURE 12-7. Serological profile, including temporal relationships integrated with infectious status and symptoms, in 75% to 85% of patients with acute type B hepatitis. (Adapted with permission from educational material of Abbott Laboratories, North Chicago, IL.)
In addition to serological tests, sensitive molecular assays may be used to detect HBV DNA, revealing active viral replication in either acute or chronic infection.55 These assays may be useful for early detection, as in screening blood donors, since DNA is detectible an average of 25 days before seroconversion.49 Additionally, some assays allow the quantification of serum viral load, which may be used in the decision to treat and, subsequently, monitor therapy. Presently eight genotypes of HBV have been identified and identification of these genotypes can be of real value in terms of determining appropriate therapy for chronic infection. For example genotype A, most prevalent in the United States, seems to respond better to interferon than the others, and patients with this genotype might benefit from starting with interferon as opposed to the oral agents available. Genotype C is more prevalent in Asia. For the details of these assays (PCR, RNA:DNA hybrid capture assay, nucleic acid cross-linking assay, and branched DNA assay), the reader is referred to a recent review by Pawlotsky et al.56
Acute Type B Hepatitis
HBsAg titers usually develop within 4–12 weeks of infection and may be seen even before elevation of aminotransferases or clinical symptoms (see Figure 12-7). Subsequently, HBsAg levels decline as anti-HBs titers develop, which indicates resolution of the acute symptomatic infection and development of immunity. In between the decline of HBsAg and the rise of anti-HBs, there is often a window when neither is present during which time anti-HBcAg may be used to diagnose infection. IgM anti-HBcAg may be used to reveal acute infection as opposed to a flare of chronic HBV.55
Chronic hepatitis is defined as persistently elevated LFTs for 6 months. The development of chronic hepatitis B is suggested by the persistence of elevated LFTs (aminotransferases) and is supported by persistence of HBsAg for more than 6 months after acute infection. Persistence of HBeAg also suggests chronic infection, but some chronically infected patients produce anti-HBeAg and, subsequently, clear HBeAg well after the acute phase is over (late seroconversion; see Figure 12-8). Clearance of the HBeAg is associated with a decrease in viral DNA, and some degree of remission in chronic hepatitis B. This, however, can be confusing as HBeAg is a precore protein, and in patients infected with certain mutations developed during the course of the disease (precore and core promoter) HBeAg may not be produced. Yet even in the face of anti-HBe, there may be active disease with ongoing fibrosis and development of cirrhosis. Although chronically infected individuals usually lack anti-HBsAg, in some cases low levels of non-neutralizing antibodies may be present. Additionally, low levels of IgM anti-HBcAg may persist.48
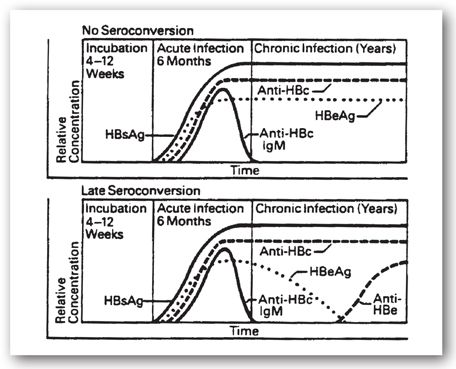
FIGURE 12-8. Serological profiles of patients who chronically carry hepatitis B virus. (Reproduced with permission from Abbott Laboratories, North Chicago, IL.)
Hepatitis B Vaccine
The HBV vaccine consists of recombinant HBsAg, which is not infectious, and it stimulates the production of protective anti-HBs. Generally, this is a safe vaccine, with efficacy of over 90%. It is indicated for people at high risk of acquiring type B hepatitis or its complications including neonates of mothers with hepatitis B, men who have sex with men, injection drug abusers, dialysis patients, healthcare workers, HIV patients, family and household contacts of patients with type B hepatitis, sexually active people with multiple partners, and patients with chronic liver disease. More recent efforts, especially in endemic countries, are leading to this being accepted as a universal vaccine. This vaccine, for example, is often required for students in the United States before entering public school. A vaccine product directed against both HAV and HBV is available. In analyzing serologic data, successful vaccination may be distinguished from previous infection by the presence of anti-HBs and absence of antibodies against other antigens (e.g., HBcAg, HBeAg). Testing for antibodies after vaccination is not generally recommended, with exceptions including healthcare workers, dialysis patients, spouses or sexual partners of infected patients. If these individuals test negative for the antibody to HBV, they should receive a second series of vaccine doses. While some patients will fail to develop antibodies for a number of reasons, including anergy, if antibody tests are negative after the second vaccine series the patient should be evaluated for the possibility of occult HBV infection. The vaccine has a prolonged duration of action. Routine booster injections are not recommended except, perhaps, for dialysis patients when their titers of anti-HBs are less than 10 International Units/L.
Hepatitis C virus (HCV) is an RNA virus mainly spread parenterally, although it may also be transmitted vertically and sexually.54 While 70% to 80% of acute infections are asymptomatic, 70% to 80% of patients develop chronic disease.49 Given the mildness of the acute attack and the tendency to develop into chronic hepatitis, it is understandable why many patients with this disease first present decades later with cirrhosis or more commonly chronic elevations of aminotransferases. Because chronic HCV infection is often asymptomatic and LFTs may be normal or intermittently elevated, it is recommended that patients at high risk for HCV be screened appropriately. Patients for whom screening would be appropriate include those with a history of illegal drug use, including snorting cocaine, as well as parenteral drug abuse. Additionally patients who received clotting factors before 1987 or blood products or organ transplants before July 1992, and patients with a history of hemodialysis should be screened.
Acute hepatitis C is often asymptomatic, and when symptoms are present they are mild. Diagnosis of acute hepatitis C, however, is important as evidence suggests that prompt treatment with antiviral medications can prevent progression to chronic hepatitis C in a majority of cases.
In chronic hepatitis C infection, the LFTs are usually minimally elevated with ALT and AST values commonly in the 60–100 International Units/L range. These values can fluctuate and occasionally return to normal for a year or more, only to rebound when next checked.57 The primary clinical concern in chronic HCV is that if untreated, within 20 years 20% to 30% of patients develop cirrhosis and 1% to 5% develop hepatocellular carcinoma.49
The first screening test used is often an ELISA (also referred to as anti-HCV), which detects antibodies against a cocktail of HCV antigens. Positive tests can be seen in patients who have passively acquired these antibodies (but not the infection), as in after transfusions, or children of mothers with hepatitis C. Due to possible cross-reactivity with one of the antigens in the assay, this test has a considerable false-positive rate, and thus positive results need to be confirmed with a more specific assay. One such assay is the recombinant immunoblot assay (RIBA), which is similar to ELISA in principle, but tests antibody reactivity to a panel of antigens individually. Binding to two or more antigens is considered a positive test.51 Binding to one antigen is considered indeterminate. Presently the approach to a positive ELISA is to skip the RIBA and go directly to the reverse transcriptase polymerase chain reaction (RT-PCR) assay.
Qualitative RT-PCR, often referred to as just PCR, detects viral RNA in the blood. It is a very sensitive assay that may be used in diagnosis and subsequent management of hepatitis C. RT-PCR has several advantages compared to serologic tests. It can detect HCV within 1–2 weeks of exposure and weeks before seroconversion, symptoms, or the elevation of LFTs. This may be useful because seroconversion has only occurred in 70% to 80% of patients at the onset of symptoms, and it may never occur in immunosuppressed patients.57 Additionally there is now evidence suggesting that treating acute hepatitis C may be of value. Some immunocompromised patients with hepatitis C (as above) may have false-negative ELISA studies, and thus the PCR is recommended for consideration in patients with hepatitis or chronic liver disease who are immunosuppressed. Furthermore, unlike serologic assays, RT-PCR is not confounded by passively acquired antibodies that may be present in uninfected infants or recipients of blood products, and RT-PCR can distinguish between resolved and chronic infection.
Once a diagnosis of HCV infection is established, various quantitative molecular assays that monitor viral load may be useful in following viral titers during treatment or assessing likelihood of response to therapy. A major consideration with these tests is that the methodology is not yet standardized, and there is lab-to-lab variability. These tests are not preferred for initial diagnosis since they are less sensitive than qualitative RT-PCR. They include a quantitative PCR assay and a branched-chain DNA assay (for more information, see Pawlotsky et al.).56 Presently most labs report HCV PCR measurements in International Units/mL, with pretreatment levels often in the millions.
There are at least six major genotypes of the type C virus and multiple subtypes. Viral genotype determination is useful since genotype is known to affect the likelihood of response to certain treatments. Additionally, while the treatment for the most common genotypes found in the United States (type 1) is 1 year in duration, current recommendations suggest that some other genotypes (notably type 2 or 3) can be successfully treated with only 6 months of therapy. As newer therapies become available, length of treatment may vary depending not only on genotype but on the rapidity of a patient’s response to therapy.58 Presently two protease inhibitors are approved for treatment of hepatitis C, when used in conjunction with ribavirin-interferon therapy, telaprevir and boceprevir. Genotype determination may be done via direct sequencing or hybridization of PCR amplification products.57
Hepatitis D virus (HDV) is caused by a defective virus that requires the presence of hepatitis B surface antigen (HBsAg) to cause infection. Therefore, people can only contract type D hepatitis concomitantly with HBV infection (coinfection) or if chronically infected with HBV (superinfection). Coinfection presents as an acute infection that may be more severe than HBV infection alone.49 Alternatively, the picture of superinfection is that of a patient, with known or unknown chronic HBV, who develops an acute flare with worsening liver function and increases in HBsAg.51 Acute coinfection is usually self-limited with rare development of chronic hepatitis, while superinfection becomes chronic in more than 75% of cases and increases the risk of negative sequelae such as cirrhosis. Transmission of HDV is generally by parenteral routes, although no obvious cause can be determined in some cases.
Testing for HDV is usually only indicated in known cases of HBV infection. The single, widely available assay detects anti-HDV antibodies of all isotypes (Figure 12-9). This test is unable to distinguish between acute, chronic, or resolved infection and lacks sensitivity since only about 38% of infected patients have detectible anti-HDV within the first 2 weeks of illness.55 Because seroconversion may occur as late as 3 months after infection, testing may be repeated if the clinical picture suggests HDV.48 Tests are also available for HDV RNA, and stains are available to assess the D antigen in hepatocytes.
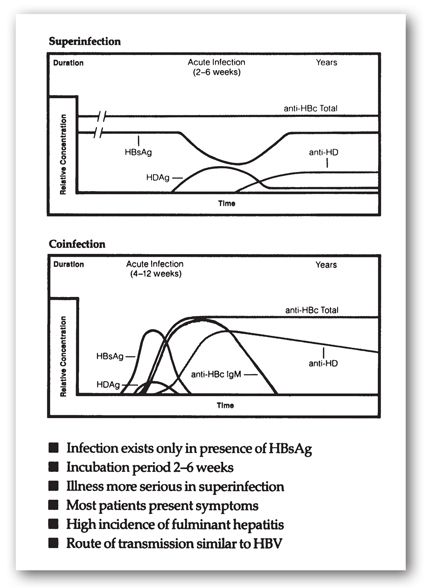
FIGURE 12-9. Two serological profiles of patients infected with the hepatitis D virus. HDAg = hepatitis D antigen (the virus); anti-HD = antibodies against hepatitis D virus. (Reproduced with permission from Abbott Laboratories, North Chicago, IL.)
Type E Hepatitis
Hepatitis E virus (HEV) is generally quite similar to hepatitis A. It is a hardy, protein-coated RNA virus that is spread via a fecal-oral route often by contaminated food or water. Like HAV, HEV causes an acute illness that is generally self-limited, and HEV is endemic in parts of Asia. It is becoming increasingly recognized in the United States. Unlike HAV, HEV is notable for a predilection for causing life-threatening illness in women who are in their third trimester of pregnancy. Recently, testing for antibodies to hepatitis E has become available, including HEVAg, or hepatitis E antigen. Work is underway to develop a vaccine for hepatitis E.
Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is a chronic disease involving progressive destruction of small intrahepatic bile ducts leading to cholestasis and progressive fibrosis over a period of decades. It can progress to cirrhosis and liver failure, necessitating transplantation. Ninety percent of affected individuals are female, with onset occurring between the early twenties and late eighties.59,60 The etiology of the disease is unknown, although it seems to involve an autoimmune component, and has associations with a variety of autoimmune disorders including Sjögren syndrome, rheumatoid arthritis, and scleroderma. The initial symptoms of the disease are often those of progressive cholestasis with fatigue, pruritus, jaundice, and deficiencies in fat-soluble vitamins.60
The most useful laboratory test in diagnosing PBC is the detection of antimitochondrial antibody (AMA), with a sensitivity of 95%.61 This assay is also highly specific, although patients with autoimmune and drug-induced hepatitis occasionally have low antibody titers. PBC usually presents with a predominantly cholestatic laboratory picture, initially with an elevated ALP and GGT, and later, with an elevated bilirubin. Aminotransferases tend to be minimally elevated or normal.
TESTS TO ASSESS PANCREATIC INFLAMMATION/PANCREATITIS
Pancreatitis describes inflammation of the pancreas (either acute or chronic) and is the most common disease associated with this gland. Although there are multiple causes of pancreatitis, the clinical presentation is often the same. Acute pancreatitis generally presents with severe midepigastric abdominal pain developing over an hour, often radiating to the back. The pain tends to be continuous and can last for several days.
This condition is often associated with nausea and vomiting; in severe cases, fever, ileus, and hypotension can occur. Ultimately, there can be progressive anemia, hypocalcemia, hypoglycemia, hypoxia, renal failure, and death. The clinician faces the challenge of rapidly establishing this diagnosis, because many conditions (e.g., ulcers, biliary disease, myocardial infarction, and intestinal ischemia or perforation) can present in a similar manner. Most individuals with acute pancreatitis can make full recoveries; however, the long-term inflammation associated with chronic pancreatitis can lead to fibrosis and calcification of the pancreas causing irreversible damage, occasionally leading to the development of diabetes mellitus or malabsorption due to deficiencies in the production of pancreatic enzymes.
Gallstones and alcohol abuse are causative factors in 60% to 80% of acute pancreatitis cases.62 Medications can also cause acute pancreatitis (Table 12-8). Other possible causes include autoimmune diseases, trauma (a typical example being an injury due to a bicycle handlebar), penetrating ulcers, hypercalcemia, hypertriglyceridemia, pancreatic neoplasm, and hereditary or autoimmune pancreatitis. Often, however, it is impossible to determine the definite cause of a patient’s attack.
a5-ASA drugs refers to 5-aminosalicylic acid agents (e.g., olsalazine, mesalamine, and sulfasalazine).
The tests discussed in this section, amylase and lipase, are primarily used to diagnose pancreatitis, although they may be clinically useful in the diagnosis of other pathologies.
Amylase
Normal range: 20–96 units/L (method dependent)
Amylase helps break starch into its individual glucose molecules. The enzyme’s most frequent clinical use is in the diagnosis of acute and chronic pancreatitis. While amylase levels are often used for this diagnosis, increasingly lipase (see below) is preferred in part due to the longer half-life of the latter.
As with any serum protein, concentrations result from the balance between entry into circulation and rate of clearance. Most circulating amylase originates from the pancreas and salivary glands. These sources are responsible for approximately 40% and 60% of serum amylase, respectively. However, the enzyme is also found in the lungs, liver, fallopian tubes, ovary, testis, small intestine, skeletal muscle, adipose tissue, thyroid, tonsils, and certain cancers, and various pathologies may increase secretion from these sources. The kidneys are responsible for about 25% of the metabolic clearance, with the remaining extrarenal mechanisms being poorly understood. The serum half-life is between 1–2 hours.63,64 Patients with azotemia can have decreased amylase clearance, and elevated amylase levels. More than half of the patients who have a creatinine clearance between 13–39 mL/min have elevated amylase levels.65 Although there is no amylase activity in neonates and only small amounts at 2–3 months of age, concentrations increase to the normal adult range by 1 year.
Amylase concentrations rise within 2–6 hours after the onset of acute pancreatitis and peak after 12–30 hours if the underlying inflammation has not recurred. In uncomplicated disease, these concentrations frequently return to normal within 3–5 days. More prolonged, mild elevations occur in up to 10% of patients with pancreatitis and may indicate ongoing pancreatic inflammation or associated complications (e.g., pancreatic pseudocyst).
Although serum amylase concentrations do not correlate with disease severity or prognosis, a higher amylase may indicate a greater likelihood that the patient has pancreatitis.66 For example, serum amylase concentrations may increase up to 25 times the upper limit of normal in acute pancreatitis while elevations from opiate-induced spasms of the sphincter of Oddi generally are less than 2–10 times the upper limit of normal. Unfortunately, the magnitude of enzyme elevation can overlap in these situations, and ranges are not very specific.
A Case of a Prolonged PT and Low Serum Albumin
JANE M., A 50-YEAR-OLD WOMAN, presented to her physician complaining of increasing fatigue and a 20-lb weight loss over the past 4 months. Initial evaluation showed an albumin of 2 g/dL (4.0–5.0 g/dL) and a PT of 18 seconds (12.7–15.4 seconds). Jane M. was referred for evaluation of possible cirrhosis. On further questioning, she denied any history of hepatitis, exposure to hepatotoxins, alcohol use, family history of liver disease, or liver disease.
Jane M.’s physical examination did not suggest liver disease; there was no evidence of ascites, palmar erythema, asterixis, hepatomegaly, splenomegaly, or spider angiomata. It was noted that she had pedal edema. Liver function studies were otherwise normal: ALT, 12 International Units/L (7–41 International Units/L); AST, 20 International Units/L (12–38 International Units/L); total bilirubin, 1 mg/dL (0.3–1.3 mg/dL); and ALP, 56 International Units/L (33–96 International Units/L).
An IM dose of vitamin K 10 mg corrected the PT (12 seconds) within 48 hours. Workup showed that Jane M. had malabsorption due to sprue, a disease of the small bowel. With proper dietary management, her symptoms resolved and she gained weight. At a followup visit 3 weeks later, her albumin concentration was 3.7 g/dL and her edema had resolved.
Question: Why did Jane M. develop a low albumin and a prolonged PT? What caused her pedal edema?
Discussion: This case demonstrates that while low albumin and a prolonged PT suggest advanced liver disease, other causes need to be considered. Administration of vitamin K promptly corrected Jane M.’s PT, suggesting malabsorption of vitamin K. If she had had cirrhosis, her PT would not have corrected with the vitamin K. Similarly, her hypoalbuminemia was not due to her liver’s inability to synthesize albumin but to the malabsorptive disorder that was interfering with protein absorption. Therefore, Jane M. had a low albumin and elevated PT in the absence of liver disease. Her pedal edema was due to hypoalbuminemia secondary to malabsorption.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


