Fig. 48.1 Major pathways in lipoprotein formation and metabolism.
Dietary lipids including cholesterol (CHOL) and free fatty acids (FFA) in the gut are emulsified by bile acids and transported within chylomicrons (CHY) to the liver. They are then circulated as cholesterol and triglycerides (TG) to tissues in very-low-density lipoproteins (VLDL), where endothelial lipoprotein lipase (LPL) liberates FFA in adipose and muscle for storage or metabolism. The resulting low-density lipoproteins (LDL) are returned to hepatocytes via LDL receptors, or taken up by LDL receptors in extrahepatic tissues where they are oxidised and contribute to atherogenesis. The high-density lipoprotein (HDL) pool (nascent HDL) is derived from the chylomicrons following the action of LPL, and the reverse cholesterol pathway returns HDL to the liver via HDL receptors. Lipid-lowering drugs and their principal targets are shown with red arrows. ACoA, acetyl coenzyme A; Apo, apolipoprotein; CERP, cholesterol efflux regulatory protein; CHY rem, chylomicron remnant; FATP, fatty acid transport protein; HMG-CoA reductase, β-hydroxyl-β-methylglutaryl-coenzyme A reductase.
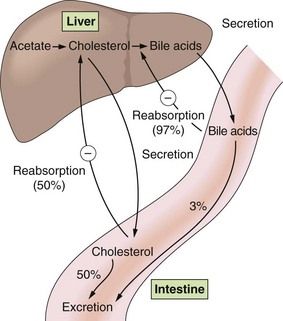
Fig. 48.2 Enterohepatic cycling of cholesterol and bile acids.
Bile acids are secreted via the bile duct into the duodenum, where they aid in dietary lipid absorption, and are then returned to the liver by the portal circulation. A circled minus sign indicates a negative-feedback effect. Percentages are in relation to the amount excreted in bile.
Triglycerides (fatty acids esterified with glycerol) are the major dietary fat, and can also be synthesised from intermediary metabolites formed in the liver from excess carbohydrate in the diet. Triglycerides are stored in adipose tissue, from where they can be mobilised as non-esterified free fatty acids to act as an energy substrate during periods of fasting.
The basis of lipoprotein metabolism
Lipids (triglycerides and esters of cholesterol) have low water solubility. They circulate in plasma encased in a coat of apolipoproteins within a phospholipid monolayer, creating lipoproteins that are water soluble and which make the triglycerides and cholesterol esters transportable. The lipoproteins can be differentiated according to the triglyceride/cholesterol ratio they carry, their apolipoprotein constituent and their density (Table 48.1). They are usually classified according to their density into very-low-density (VLDL), low-density (LDL), intermediate-density (IDL) and high-density (HDL) lipoproteins. There are specific cell-surface receptors for processing different apolipoproteins and these determine where and how particular fractions of circulating cholesterol and triglyceride will be handled (Table 48.1). In healthy individuals, about 70% of plasma cholesterol is carried by LDL and 20% by HDL. The least-dense and largest-diameter particles, known as chylomicrons, are exclusively concerned with the transport of dietary lipid from the intestine to the liver. Their low density and large size reflect their high content of triglycerides (Table 48.1), and they are almost completely removed from blood after a 12 h fast. VLDL carries about 60% of plasma triglyceride in the fasting state. The ratio of cholesterol to triglyceride carried is greatest in the HDL fraction.
Table 48.1
Apolipoprotein and lipid composition of major lipoproteins and their sources
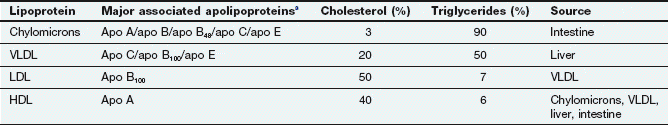
HDL, high-density lipoproteins; LDL, low-density lipoproteins; VLDL, very-low-density lipoproteins.
aThe apolipoproteins supply structural integrity and also have roles in controlling lipoprotein metabolism and as receptor ligands.
Processing of lipids absorbed from the gut
Cholesterol and free fatty acids are solubilised by bile acids in the gut lumen to facilitate absorption into enterocytes (see Fig. 48.1). Soluble cholesterol is transported into the enterocyte from the intestinal lumen by a specific lipid transmembrane transporter called Nieman-Pick C1-like 1 protein (NPC1L1). Triglyceride absorption does not require a specific transporter. Cholesterol and triglycerides are then incorporated into chylomicrons within the enterocytes (see Table 48.1). Chylomicrons pass into the lymphatic system and then into the circulation. Hydrolysis of triglycerides to free fatty acids in the chylomicrons is carried out by a lipoprotein lipase attached to the endothelium of capillaries in muscle and adipose tissue, and requires the chylomicron-associated apolipoprotein C (subtype C2) as a cofactor (Table 48.1). Free fatty acids are utilised by muscle and liver as an energy source or stored as triglycerides in adipose tissue. After removal of triglycerides from the chylomicrons, the remaining surface lipoprotein and lipid fractions leave the particles to enter the HDL pool as ‘nascent HDL’ (Fig. 48.1). The chylomicron remnants are taken into hepatocytes by specific chylomicron remnant (apolipoprotein E, or apo E) receptors.
Plasma transport and liver processing of lipids
Cholesterol is transported to tissues in chylomicrons, VLDL and LDL; it is transported from tissues to the liver by HDL. A high plasma concentration of LDL is associated with atheromatous disease.
Liver cholesterol (as esters) and any triglycerides in the liver that are surplus to synthetic and oxidative requirements are released into the circulation complexed to VLDL. Peripheral lipoprotein lipase acts on VLDL to release free fatty acids, leaving IDL (not shown in Fig. 48.1); triglycerides in IDL are also hydrolysed by hepatic lipase to release free fatty acids, which generates LDL. LDL therefore contains a higher concentration of cholesterol and a lower concentration of triglyceride compared with VLDL (Table 48.1). LDL is removed from the circulation by uptake into liver cells (75%) and peripheral tissues (25%). Some 70% of this uptake is by specific receptors for the apolipoproteins type B and E (Fig. 48.1), while the rest is by non-receptor-mediated pathways. The circulating concentration of LDL rises if there is either excess production of LDL or deficient LDL receptor numbers. When plasma LDL rises, non-receptor-mediated uptake of cholesterol in peripheral tissues such as arterial walls will increase. Within arterial walls, LDL cholesterol undergoes oxidation which leads to formation of lipid-rich deposits and atheromatous plaques (see below).
HDL carries cholesterol mobilised from peripheral tissues (and particularly oxidized cholesterol derivatives), and transports it to the liver (reverse cholesterol transport). The efflux of cholesterol from peripheral cells is facilitated by cholesterol efflux regulatory protein (CERP). This cholesterol is bound to nascent HDL, and then the cholesterol is esterified by the circulating enzyme lecithin-cholesterol acyltransferase (LCAT) to create mature HDL. HDL is believed to protect against atheroma by this reverse cholesterol transport from peripheral tissues to the liver. The enzyme cholesterol ester transfer protein (CETP) can transfer cholesterol from HDL to VLDL in exchange for triglycerides. The extent of this exchange depends on the concentration of circulating triglycerides.
Hypercholesterolaemia and atheroma
Abnormalities of plasma lipoprotein metabolism produce excessive concentrations of circulating cholesterol and/or triglyceride. Their clinical importance lies in their relationship to the production of atheroma (mainly raised plasma LDL cholesterol with a contribution from triglycerides) and pancreatitis (plasma triglycerides >12 mmol⋅L−1). Atheroma is focal thickening of the intima of arteries, produced by a combination of cells, elements of connective tissue, lipids and debris.
Excess LDL accumulates in the arterial wall, where its cholesterol undergoes enzymatic and free radical oxidation to produce a cytotoxic and chemotactic lipid that can activate the endothelium. Activated endothelium (a state which is also initiated by other atherogenic factors such as smoking, diabetes or hypertension) expresses adhesion molecules that attract platelets, monocytes and some T-lymphocytes. These cells migrate into the sub-endothelial space, where the monocytes differentiate into macrophages under the influence of endothelial cytokines. The macrophages take up oxidised LDL cholesterol via scavenger receptors, and the cholesterol accumulates as droplets in the cytosol, creating lipid-rich foam cells. Foam cells initiate fatty streaks that are the precursor of atheroma. T-cells in the developing atheromatous lesion recognise lipid antigens and release various cytokines that attract further inflammatory cells and initiate a T-helper cell type 1 inflammatory response (Ch. 38). These processes also result in formation of a cap of smooth muscle cells and collagen-rich matrix over the lesion. The extent of the inflammatory response determines whether the cap becomes fibrous and stable, or is destabilised by infiltration of inflammatory cells that make the cap prone to rupture or surface erosion. Plaque destabilisation underlies the development of acute coronary syndromes and many cases of ischaemic stroke (see Chs 5 and 9).
Atherogenic patterns of lipoproteins can result from the following:

 primary (inherited) disorders of enzymes or receptors involved in lipid metabolism. Most inherited hyperlipidaemias are polygenic, but an important inherited defect is familial hypercholesterolaemia, a single recessive gene disorder that affects 1 in 500 of the population, who have reduced synthesis of LDL receptors,
primary (inherited) disorders of enzymes or receptors involved in lipid metabolism. Most inherited hyperlipidaemias are polygenic, but an important inherited defect is familial hypercholesterolaemia, a single recessive gene disorder that affects 1 in 500 of the population, who have reduced synthesis of LDL receptors,
A classification for the various phenotypic patterns of primary hyperlipidaemia adopted by the World Health Organization is shown in Table 48.2.
Table 48.2
The Fredrickson classification of dyslipidaemias
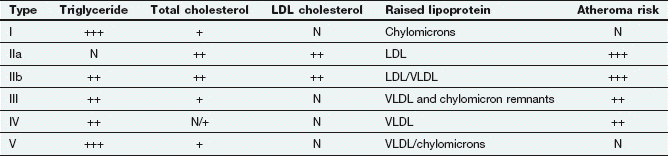
N, normal; +, slightly raised; ++, moderately raised; +++, extremely raised.
Drugs for hyperlipidaemias
HMG-CoA reductase inhibitors (‘statins’)
Mechanism of action and effects: HMG-CoA reductase inhibitors competitively inhibit the enzyme that catalyses the rate-limiting step in the synthesis of cholesterol (Fig. 48.1). Their most important action is in the liver, where the fall in hepatic cholesterol levels produces a compensatory upregulation in the number of LDL receptors on hepatocytes, with increased clearance of circulating LDL cholesterol. In the liver, the cholesterol is reprocessed to form bile salts. The extent of the reduction in plasma LDL cholesterol depends on the specific drug and the dose of the drug ranges from 25 to 50%. Short-acting statins, such as simvastatin, are most effective when taken at night, which is the time when most cholesterol synthesis occurs. Statins also reduce the circulating concentration of VLDL by stimulating lipoprotein lipase, and therefore reduce circulating triglycerides. A modest increase in HDL cholesterol is usually seen, due to increased synthesis of the constituent apolipoprotein A1. This is due to activation of peroxisome proliferator-activated receptor α (PPAR-α) (see also fibrates, below).
Statins have several other actions, which may be distinct from their ability to reduce plasma lipids (Box 48.1). There is increasing evidence that some of these may contribute significantly to the beneficial actions of statins in reducing clinical events in people with atherothrombotic disease.





Pharmacokinetics: The statins are well absorbed from the gut. Simvastatin is a prodrug (Ch. 2) that is activated in first-pass metabolism in the liver by cytochrome P450 (CYP3A4). Further metabolism inactivates the drug and only 5% of the active compound reaches the circulation. Atorvastatin undergoes first-pass metabolism, in part to active derivatives, and has a very long half-life. Pravastatin is a hydrophilic drug that is eliminated mainly by the kidneys; its half-life is 1–2 h. Rosuvastatin has a low oral bioavailability and is eliminated mainly in the bile, with a half-life of 20 h.



 Myalgia (muscle pain) or myositis (muscle inflammation) and rarely rhabdomyolysis. In some people there is a raised serum creatine kinase concentration suggesting muscle damage, but no muscle symptoms. The mechanism of the myopathy remains uncertain but it is likely that some people have minor metabolic abnormalities in their striated muscle that makes the muscle more susceptible to reduction of its fat substrate. There is an increased risk when a statin is used in combination with a fibrate, nicotinic acid, fusidic acid, ciclosporin and several other drugs (Ch. 38).
Myalgia (muscle pain) or myositis (muscle inflammation) and rarely rhabdomyolysis. In some people there is a raised serum creatine kinase concentration suggesting muscle damage, but no muscle symptoms. The mechanism of the myopathy remains uncertain but it is likely that some people have minor metabolic abnormalities in their striated muscle that makes the muscle more susceptible to reduction of its fat substrate. There is an increased risk when a statin is used in combination with a fibrate, nicotinic acid, fusidic acid, ciclosporin and several other drugs (Ch. 38).Specific cholesterol absorption inhibitors
Mechanism of action: Ezetimibe acts at the brush border of the small intestinal mucosa to specifically inhibit the NPC1L1 transporter and reduces cholesterol absorption by about 50%. It has no effect on the absorption of triglycerides, bile acids or fat-soluble vitamins. Given alone, ezetimibe reduces plasma total cholesterol by about 15% and LDL cholesterol by about 20%. When taken with a low dose of statin the combination is as effective as three doublings of the statin dose in reducing plasma total cholesterol.
Pharmacokinetics: Ezetimibe is rapidly but incompletely absorbed from the gut, and metabolised in the gut wall and the liver. It undergoes enterohepatic circulation, which gives it a long half-life (about 22 h), and about 80% is excreted in the faeces.



Bile acid-binding (anion-exchange) resins
Mechanism of action: Bile acids are synthesised from cholesterol in the liver, and are secreted into the duodenum to aid absorption of dietary fat. They are then reabsorbed in the terminal ileum and returned to the liver in the portal circulation (Fig. 48.2). Bile acid-binding resins are insoluble, non-absorbable polymers that bind bile salts in the gut and prevent enterohepatic circulation of bile acids.
When reabsorption of bile acids is impaired by binding to the resin, bile acid synthesis is increased from cholesterol in the liver. This reduces intrahepatic cholesterol, and there is compensatory upregulation of hepatic LDL receptors in order to replenish liver cholesterol. LDL cholesterol is cleared more rapidly from plasma, with a fall in circulating levels of 15–20%. Stimulation of VLDL synthesis produces a small rise in plasma triglycerides. There is a small rise in HDL cholesterol, but the mechanism for this is unclear.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


