Fig. 5.1 Aspects of inflammatory processes that contribute to coronary heart disease.
Multifactorial processes contribute to coronary heart disease; endothelium is damaged and activated; platelets adhere and promote leucocyte infiltration and thrombus formation; low-density lipoprotein (LDL) is oxidised and taken up via scavenger receptors into monocyte-macrophages, subsequently forming foam cells. Dysfunctional expression of a host of cytokines, lipid mediators, free radicals and proteases exacerbates inflammation, endothelial damage, atheroma formation, plaque rupture and thrombus formation. These processes are influenced by risk factors such as smoking, heredity, hypercholesterolaemia, hypertension, obesity, diabetes, age and gender. IFN-γ, interferon-γ; IL-10, interleukin-10; MMPs, matrix metalloproteases; TGF-β, tumour growth factor β; Th, T-helper cell; VCAM-1, vascular cell adhesion molecule 1.
Atheromatous plaques tend to form in areas of flow disturbance, such as bends in the vessels or near branching vessels. The major risk factors for atheromatous coronary artery disease (in common with atheroma in other parts of the vascular tree) are male sex, smoking, hypertension, hypercholesterolaemia and diabetes mellitus. The effects of these risk factors are additive, and when several are present coronary atheroma occurs more extensively and at a younger age.
Early atheromatous plaques enlarge by stretching the medial smooth muscle (remodelling) and do not narrow the lumen of the vessel until 40–50% of the cross-sectional area of the vessel is diseased. Even when luminal narrowing occurs, symptoms only arise when 75% of the cross-sectional area of the vessel lumen is occluded.
Although atheroma can diffusely involve a long segment of the vessel, plaques are often confined to a small segment of the coronary artery. Localised plaques frequently involve only part of the circumference of the arterial wall, leaving the rest free of significant disease and still able to respond to vasoconstrictor and vasodilator influences. At the site of an atheromatous plaque there is turbulent blood flow. The consequent changes in shear stress at the endothelial surface impair endothelial function and reduce local generation of vasodilator substances such as nitric oxide (see organic nitrates below). Therefore, diseased segments of an artery are particularly prone to vasospasm, which produces dynamic flow limitation superimposed on the fixed atheromatous narrowing. If the coronary artery disease is long-standing, then collateral vessels can develop around the atheromatous narrowing and improve perfusion distal to the diseased segment of the artery.
There are two morphological types of atheromatous plaque. Some have a lipid-rich core, with a substantial infiltration of inflammatory cells and a thin fibrous cap. Such plaques are relatively unstable (‘vulnerable’ plaques) and are more prone to plaque disruption by ulceration or rupture of the cap, leading to thrombus formation (see below). Other plaques have a fibrotic core, with a thick fibrous cap, and are more stable. The reasons why both stable and unstable plaques can coexist in the coronary circulation is not well understood.
Reversible myocardial ischaemia is the consequence of an imbalance between oxygen supply and oxygen demand in a part of the myocardium (Fig. 5.2) due to an inability to increase coronary blood flow sufficiently to meet the metabolic demands of the heart. Rupture of an atheromatous plaque is responsible for most acute ischaemic cardiac events (presenting as an acute coronary syndrome).
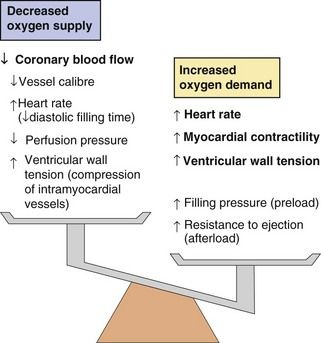
Fig. 5.2 Factors increasing myocardial oxygen demand and decreasing myocardial oxygen supply in angina.
Anti-anginal drugs act at many different sites to reduce oxygen demand and increase oxygen supply.
Clinical manifestations of myocardial ischaemia
Angina pectoris is pain arising from heart muscle after it switches to anaerobic metabolism, and is a symptom of reversible myocardial ischaemia. Ischaemia occurs once the coronary artery lumen is narrowed sufficiently to restrict maximal blood flow to a level that cannot deliver adequate oxygen to meet the metabolic needs of the myocardium. Stable angina is relatively predictable ischaemic chest pain that is most frequently experienced as chest pain on exertion or with emotional stress and is rapidly relieved by rest. Reversible myocardial ischaemia can also present with shortness of breath (due to diastolic stiffening of the left ventricle when a reduced cellular energy supply impairs the uptake of Ca2+ by the sarcoplasmic reticulum; see also heart failure with preserved ejection fraction, Ch. 7), or it can occur without symptoms (silent ischaemia). Vasospasm at the site of an atheromatous plaque accentuates the reduction in flow produced by a fixed atheromatous obstruction, and when it is present angina occurs at a lower workload.
People with stable angina have an increased risk of subsequent myocardial infarction or sudden cardiac death, due to rupture of an unstable atheromatous plaque (see below). On average the annual rate of such events is about 2%.
Acute coronary syndromes (unstable angina, myocardial infarction and sudden death)
Acute coronary syndromes have a common pathophysiological origin, arising from disruption of an unstable atheromatous plaque (vulnerable plaque) in a coronary artery. Plaque disruption can be precipitated by sudden stresses on the cap produced by pulsatile blood flow across the plaque, by elastic recoil of the vessel in diastole or by vasospasm. As a consequence of these stresses the thin cap over the plaque fissures or ulcerates, leading to plaque rupture and exposure of the core of the plaque to circulating blood. Plaque rupture initiates platelet adhesion and then aggregation (Ch. 11), followed by thrombus formation and local vasospasm. These processes lead to a sudden reduction in blood flow. Platelet–thrombin microemboli can break off from the thrombus and become lodged in small distal vessels downstream from the thrombus, contributing to ischaemia.
Unstable angina
Unstable angina occurs if there is incomplete occlusion of the coronary artery following plaque rupture, but with critical reduction in blood flow so that oxygen supply is inadequate at rest or on minimal stress. Angina may then occur at rest or with very little exertion. Unstable angina is distinguished pathologically from other acute coronary syndromes because perfusion of the ischaemic tissue remains sufficient to prevent necrosis of myocytes. Unlike myocardial infarction, symptoms of unstable angina are usually relieved by glyceryl trinitrate (see below), or resolve spontaneously within 30 min.
Following an episode of unstable angina the thrombus may become incorporated into the plaque so that after healing the plaque is substantially larger, leading to greater long-term luminal narrowing.
Myocardial infarction and sudden cardiac death
Myocardial infarction most commonly arises from complete coronary artery occlusion following disruption of an unstable atheromatous plaque. Occlusion often occurs at the site of an atheromatous lesion that previously was only producing minor or moderate stenosis of the artery and may not have caused symptoms prior to disruption. Muscle necrosis begins when the occlusion lasts for longer than 20–30 min.
Myocardial infarction is usually associated with intense, prolonged chest pain and sympathetic nervous stimulation which increases cardiac work. However, about 15% of infarctions do not present with pain, and may go unrecognised (silent infarction). The diagnosis of acute myocardial infarction requires a rise in the plasma concentrations of sensitive biochemical markers, such as cardiac-specific myoglobin or troponin, which are released from necrotic myocytes. Cell death begins in the subendocardial muscle which is furthest from the epicardial blood supply (the endocardium receives its oxygen from the ventricular cavity), and, unless perfusion is restored, it progressively extends across the full thickness of the myocardium (transmurally) over the next few hours. Activation of endogenous fibrinolysis (Ch. 11) and the presence of a good collateral circulation are factors that favour reperfusion of the ischaemic area and naturally limit the size of the infarct. If very early reperfusion occurs the damage is usually confined to the subendocardial myocardium.
A full-thickness (or transmural) myocardial infarction often produces characteristic changes on the electrocardiograph (ECG), with early ST-segment elevation and eventually pathological Q waves. The resulting infarction is referred to as an ST-elevation myocardial infarction (STEMI). A subendocardial infarction often presents without diagnostic ECG changes. In these cases the ECG may show ST-segment depression or T-wave inversion (consistent with myocardial ischaemia), or even be normal. The resulting infarction is classified as a non-ST-elevation myocardial infarction (NSTEMI), because of the absence of the characteristic ST-segment changes usually found with more extensive myocardial damage.
Myocardial infarction principally affects left ventricular muscle, and the amount of muscle lost correlates well with both early and late survival. Infarction of the anterior muscle of the left ventricle (usually resulting from an occlusion in the left coronary artery system) causes greater myocardial loss than does inferior infarction of the ventricle (usually from right coronary artery occlusion). The amount of muscle loss also determines the extent of left ventricular remodelling (a geometrical change in the left ventricle that begins with healing of the infarct), which determines the risk of subsequent heart failure.
Sudden cardiac death results when fatal ventricular arrhythmias arise from ischaemic tissue, or from ventricular rupture.
Drug treatment of angina
Drug treatment for angina is directed either:


Drugs can be taken to relieve the ischaemia rapidly during an acute attack or as regular prophylaxis to reduce the risk of subsequent episodes. Several classes of drug are used to treat angina.
Organic nitrates
Mechanism of action and effects
The organic nitrates are vasodilators that relax vascular smooth muscle by mimicking the effects of endogenous nitric oxide (NO). Enzymatic degradation of the nitrate releases NO, which combines with thiol groups in vascular endothelium to form nitrosothiols. Nitrosothiols activate guanylyl cyclase, which generates the second messenger cyclic guanosine monophosphate (cGMP; Fig. 5.3). cGMP activates protein kinase G, which reduces the availability of intracellular Ca2+ to the contractile mechanism of vascular smooth muscle, causing relaxation and vasodilation. Vasodilation is produced in three main vascular beds.
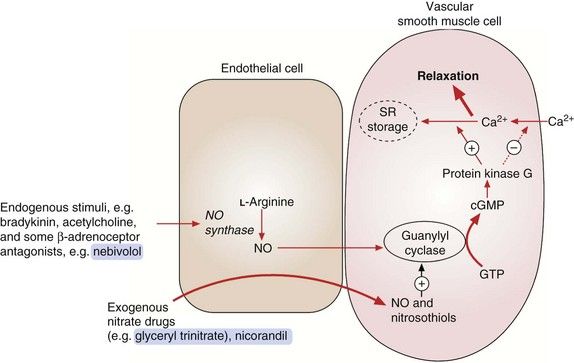
Fig. 5.3 Actions of endogenous nitric oxide (NO) and exogenous nitrates.
Endogenous NO from endothelial cells relaxes vascular smooth muscle by activating guanylyl cyclase with subsequent formation of cGMP. This activates protein kinase G, which decreases Ca2+ influx into the cell, increases Ca2+ storage in the sarcoplasmic reticulum (SR) and increases myosin light-chain dephosphorylation. Exogenous agents such as organic nitrates and nicorandil react with tissue thiols, generating NO or nitrosothiols, which then activate guanylyl cyclase and increase cGMP.
 Venous capacitance vessels, leading to peripheral pooling of blood and reduced venous return to the heart. This lowers left ventricular filling pressure (preload), decreases ventricular wall tension and therefore reduces myocardial oxygen demand. Venous dilation is produced at moderate plasma nitrate concentrations, and tolerance to this action occurs rapidly during continued treatment.
Venous capacitance vessels, leading to peripheral pooling of blood and reduced venous return to the heart. This lowers left ventricular filling pressure (preload), decreases ventricular wall tension and therefore reduces myocardial oxygen demand. Venous dilation is produced at moderate plasma nitrate concentrations, and tolerance to this action occurs rapidly during continued treatment. Arterial resistance vessels, leading to reduced resistance to left ventricular emptying (afterload). This lowers blood pressure, decreases cardiac work and contributes to a reduced myocardial oxygen demand. Arterial dilation occurs at higher plasma nitrate concentrations than venodilation, but tolerance arises less readily during long-term treatment.
Arterial resistance vessels, leading to reduced resistance to left ventricular emptying (afterload). This lowers blood pressure, decreases cardiac work and contributes to a reduced myocardial oxygen demand. Arterial dilation occurs at higher plasma nitrate concentrations than venodilation, but tolerance arises less readily during long-term treatment. Coronary arteries. Nitrates have little effect on total coronary blood flow in angina; indeed, flow may be reduced because of a decrease in perfusion pressure. However, blood flow through collateral vessels may be improved, and nitrates also relieve coronary artery vasospasm. The net effect is increased blood supply to ischaemic areas of the myocardium. Coronary artery dilation occurs at low plasma nitrate concentrations, and tolerance is slow to develop.
Coronary arteries. Nitrates have little effect on total coronary blood flow in angina; indeed, flow may be reduced because of a decrease in perfusion pressure. However, blood flow through collateral vessels may be improved, and nitrates also relieve coronary artery vasospasm. The net effect is increased blood supply to ischaemic areas of the myocardium. Coronary artery dilation occurs at low plasma nitrate concentrations, and tolerance is slow to develop.Pharmacokinetics
Glyceryl trinitrate (GTN) is the most widely used organic nitrate. It is well absorbed from the gut but undergoes extensive first-pass metabolism in the liver to inactive metabolites. To increase its bioavailability, GTN is given by one of four routes that avoid first-pass metabolism.
 Sublingual: the tablet is placed under the tongue and is absorbed rapidly across the buccal mucosa. The very short half-life of GTN (less than 5 min) limits the duration of action to approximately 30 min. Tablets lose their potency with prolonged storage, and a metered-dose aerosol spray is a more stable delivery method.
Sublingual: the tablet is placed under the tongue and is absorbed rapidly across the buccal mucosa. The very short half-life of GTN (less than 5 min) limits the duration of action to approximately 30 min. Tablets lose their potency with prolonged storage, and a metered-dose aerosol spray is a more stable delivery method. Buccal: a tablet containing GTN in an inert polymer matrix is held between the upper lip and gum, which permits slow release of drug to prolong the duration of action.
Buccal: a tablet containing GTN in an inert polymer matrix is held between the upper lip and gum, which permits slow release of drug to prolong the duration of action. Transdermal: GTN is absorbed well through the skin and can be delivered from an adhesive patch via a rate-limiting membrane or matrix. Steady release of the drug maintains a stable blood concentration for at least 24 h after application of the patch.
Transdermal: GTN is absorbed well through the skin and can be delivered from an adhesive patch via a rate-limiting membrane or matrix. Steady release of the drug maintains a stable blood concentration for at least 24 h after application of the patch.
Isosorbide 5-mononitrate is well absorbed from the gut and does not undergo first-pass metabolism. It has a half-life of 3–7 h so modified-release formulations are often used to prolong the duration of action.
Unwanted effects
 Venodilation can produce postural hypotension, dizziness, syncope and reflex tachycardia. Tachycardia can be reduced by concurrent use of a β-adrenoceptor antagonist.
Venodilation can produce postural hypotension, dizziness, syncope and reflex tachycardia. Tachycardia can be reduced by concurrent use of a β-adrenoceptor antagonist. Arterial dilation causes throbbing headaches and flushing, but tolerance to these effects is common during treatment with long-acting nitrates.
Arterial dilation causes throbbing headaches and flushing, but tolerance to these effects is common during treatment with long-acting nitrates. Tolerance to the therapeutic effects of nitrates develops rapidly if there is a sustained high plasma nitrate concentration. Tolerance is therefore a particular problem with delivery of GTN via transdermal patches or with long-acting nitrates. The cause is incompletely understood, but an important mechanism may be increased degradation of NO by oxygen free radicals (e.g. superoxides). There is limited evidence that co-administration of an angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor antagonist or hydralazine (Ch. 6) may reduce nitrate tolerance by impairing superoxide formation. Reflex activation of the sympathetic nervous system and the renin–angiotensin system in response to hypotension may also counteract the vasodilator actions of the nitrates. Tolerance can be avoided by a ‘nitrate-low’ period of several hours in each 24 h. This is preferable to a ‘nitrate-free’ period, which carries a risk of rebound angina. A nitrate-low period is achieved by asymmetric dosing with conventional formulations of isosorbide mononitrate (e.g. twice daily, at 8 a.m. and 1 p.m.) or by using a once-daily formulation that allows plasma nitrate concentrations to fall overnight. Transdermal GTN patches must be removed for part of each 24 h (e.g. overnight) to prevent tolerance.
Tolerance to the therapeutic effects of nitrates develops rapidly if there is a sustained high plasma nitrate concentration. Tolerance is therefore a particular problem with delivery of GTN via transdermal patches or with long-acting nitrates. The cause is incompletely understood, but an important mechanism may be increased degradation of NO by oxygen free radicals (e.g. superoxides). There is limited evidence that co-administration of an angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor antagonist or hydralazine (Ch. 6) may reduce nitrate tolerance by impairing superoxide formation. Reflex activation of the sympathetic nervous system and the renin–angiotensin system in response to hypotension may also counteract the vasodilator actions of the nitrates. Tolerance can be avoided by a ‘nitrate-low’ period of several hours in each 24 h. This is preferable to a ‘nitrate-free’ period, which carries a risk of rebound angina. A nitrate-low period is achieved by asymmetric dosing with conventional formulations of isosorbide mononitrate (e.g. twice daily, at 8 a.m. and 1 p.m.) or by using a once-daily formulation that allows plasma nitrate concentrations to fall overnight. Transdermal GTN patches must be removed for part of each 24 h (e.g. overnight) to prevent tolerance. Drug interactions are most troublesome with phosphodiesterase inhibitors, such as sildenafil, used in the treatment of erectile dysfunction. These inhibit cGMP metabolism (Ch. 16) and co-administration can result in marked hypotension.
Drug interactions are most troublesome with phosphodiesterase inhibitors, such as sildenafil, used in the treatment of erectile dysfunction. These inhibit cGMP metabolism (Ch. 16) and co-administration can result in marked hypotension.Beta-adrenoceptor antagonists (β-blockers)
Mechanism of action and effects in angina
All β-adrenoceptor antagonists (often simply referred to as β-blockers) act as competitive antagonists of catecholamines at β-adrenoceptors. They achieve their therapeutic effect in angina by blockade of the cardiac β1-adrenoceptor with reduced generation of intracellular cAMP. As a result they:
 decrease heart rate (by inhibition of the cardiac If pacemaker current in the sinoatrial node; see Ch. 8); this is most marked during exercise, when the rate of rise in heart rate is blunted,
decrease heart rate (by inhibition of the cardiac If pacemaker current in the sinoatrial node; see Ch. 8); this is most marked during exercise, when the rate of rise in heart rate is blunted,

The overall effect is to reduce myocardial oxygen demand. The slower heart rate also lengthens diastole and gives more time for coronary perfusion, which effectively improves myocardial oxygen supply.
Certain β-adrenoceptor antagonists have additional properties, which might reduce the incidence of unwanted effects or enhance their blood pressure-lowering actions (see below and also Chs 6 and 8), as follows.
 Cardioselectivity. Some β-adrenoceptor antagonists, for example atenolol, bisoprolol and metoprolol, are selective antagonists at the β1-adrenoceptor. They are usually called cardioselective drugs since the most important site of action on β1-adrenoceptors is the heart. Other β-adrenoceptor antagonists, for example propranolol, have equal or greater antagonist activity at β2-adrenoceptors; these drugs are referred to as ‘non-selective’ β-adrenoceptor antagonists. The cardioselectivity of all β-adrenoceptor antagonists is dose-related, with progressively more β2-adrenoceptor blockade at higher doses.
Cardioselectivity. Some β-adrenoceptor antagonists, for example atenolol, bisoprolol and metoprolol, are selective antagonists at the β1-adrenoceptor. They are usually called cardioselective drugs since the most important site of action on β1-adrenoceptors is the heart. Other β-adrenoceptor antagonists, for example propranolol, have equal or greater antagonist activity at β2-adrenoceptors; these drugs are referred to as ‘non-selective’ β-adrenoceptor antagonists. The cardioselectivity of all β-adrenoceptor antagonists is dose-related, with progressively more β2-adrenoceptor blockade at higher doses. Partial agonist activity (PAA) or intrinsic sympathomimetic activity (ISA). Certain β1-adrenoceptor antagonists also act as partial agonists at either β1– or β2-adrenoceptors. For example, pindolol is a β1-adrenoceptor antagonist that also has weak agonist activity at β2-adrenoceptors, and as such it will produce vasodilation in some vascular beds (see Fig. 6.6). Drugs with PAA at the β1-adrenoceptor have less inhibitory effect on heart rate and force of contraction and may be less effective than full antagonists in the treatment of severe angina, but their PAA means they are less likely to cause a resting bradycardia. Beta-adrenoceptor antagonists with PAA are not widely used.
Partial agonist activity (PAA) or intrinsic sympathomimetic activity (ISA). Certain β1-adrenoceptor antagonists also act as partial agonists at either β1– or β2-adrenoceptors. For example, pindolol is a β1-adrenoceptor antagonist that also has weak agonist activity at β2-adrenoceptors, and as such it will produce vasodilation in some vascular beds (see Fig. 6.6). Drugs with PAA at the β1-adrenoceptor have less inhibitory effect on heart rate and force of contraction and may be less effective than full antagonists in the treatment of severe angina, but their PAA means they are less likely to cause a resting bradycardia. Beta-adrenoceptor antagonists with PAA are not widely used. Vasodilator activity. Pure β1-adrenoceptor antagonists do not cause vasodilation. Indeed, the reflex response to β1-adrenoceptor blockade is vasoconstriction, mediated in part by the reflex sympathetic nervous system stimulation of α1-adrenoceptors in response to the fall in cardiac output. However, some β-adrenoceptor antagonists have additional properties that produce arterial vasodilation. Mechanisms of vasodilation include β2-adrenoceptor partial agonist activity (e.g. pindolol; see above), α1-adrenoceptor blockade (e.g. carvedilol, labetalol) or an increase in endothelial NO synthesis (e.g. nebivolol) (see Fig. 6.6). Vasodilation does not have any proven advantage for the treatment of angina, but may be useful when β-adrenoceptor antagonists are given for the treatment of hypertension (Ch. 6).
Vasodilator activity. Pure β1-adrenoceptor antagonists do not cause vasodilation. Indeed, the reflex response to β1-adrenoceptor blockade is vasoconstriction, mediated in part by the reflex sympathetic nervous system stimulation of α1-adrenoceptors in response to the fall in cardiac output. However, some β-adrenoceptor antagonists have additional properties that produce arterial vasodilation. Mechanisms of vasodilation include β2-adrenoceptor partial agonist activity (e.g. pindolol; see above), α1-adrenoceptor blockade (e.g. carvedilol, labetalol) or an increase in endothelial NO synthesis (e.g. nebivolol) (see Fig. 6.6). Vasodilation does not have any proven advantage for the treatment of angina, but may be useful when β-adrenoceptor antagonists are given for the treatment of hypertension (Ch. 6).Pharmacokinetics
Highly lipophilic β-adrenoceptor antagonists, such as propranolol and metoprolol, are well absorbed from the gut but undergo extensive first-pass metabolism in the liver, with considerable variability among individuals. Reduction in heart rate during exercise is closely related to the plasma concentration of the drug, so dose titration of lipophilic β-adrenoceptor antagonists is usually necessary to achieve an optimal clinical response. Most lipophilic β-adrenoceptor antagonists have short half-lives (see Compendium of drugs used to treat ischaemic heart disease at the end of this chapter), and are often available in modified-release formulations to prolong their duration of action.
Hydrophilic β-adrenoceptor antagonists, such as atenolol, are incompletely absorbed from the gut, and are eliminated unchanged in the urine. The dose range to maintain effective plasma concentrations is narrower than for those drugs that undergo metabolism. The half-lives of hydrophilic β-adrenoceptor antagonists are usually longer than those of lipophilic drugs (see Compendium at the end of this chapter).
Unwanted effects
 Blockade of β1-adrenoceptors. A large dose of a β-adrenoceptor antagonist can precipitate acute pulmonary oedema if there is pre-existing poor left ventricular function, when high sympathetic nervous activity is necessary to maintain cardiac output. However, there is a paradox that when used at low doses with gradual-dose titration a β-adrenoceptor antagonist is part of the core therapy of heart failure (Ch. 7). A reduction in cardiac output can also impair blood supply to peripheral tissues, which can be detrimental in critical leg ischaemia (Ch. 10) or can provoke Raynaud’s phenomenon (Ch. 10). Excessive bradycardia occasionally occurs, and β-adrenoceptor antagonists should be used with caution or avoided in the presence of advanced atrioventricular conduction defect (heart block). Drugs with partial agonist activity produce less bradycardia or reduction of cardiac output.
Blockade of β1-adrenoceptors. A large dose of a β-adrenoceptor antagonist can precipitate acute pulmonary oedema if there is pre-existing poor left ventricular function, when high sympathetic nervous activity is necessary to maintain cardiac output. However, there is a paradox that when used at low doses with gradual-dose titration a β-adrenoceptor antagonist is part of the core therapy of heart failure (Ch. 7). A reduction in cardiac output can also impair blood supply to peripheral tissues, which can be detrimental in critical leg ischaemia (Ch. 10) or can provoke Raynaud’s phenomenon (Ch. 10). Excessive bradycardia occasionally occurs, and β-adrenoceptor antagonists should be used with caution or avoided in the presence of advanced atrioventricular conduction defect (heart block). Drugs with partial agonist activity produce less bradycardia or reduction of cardiac output.
 Bronchospasm can be precipitated in people with asthma (including those with chronic obstructive pulmonary disease and some bronchodilator reversibility). People who are susceptible to this problem can experience bronchospasm even with cardioselective drugs.
Bronchospasm can be precipitated in people with asthma (including those with chronic obstructive pulmonary disease and some bronchodilator reversibility). People who are susceptible to this problem can experience bronchospasm even with cardioselective drugs. Hypoglycaemia may be prolonged by non-selective β-adrenoceptor antagonists, which may be a problem in people with diabetes mellitus who are treated with insulin (Ch. 40). Gluconeogenesis, a component of the metabolic response to hypoglycaemia, is dependent upon β2-adrenoceptor stimulation in the liver. Beta-adrenoceptor antagonists also blunt the autonomic response that alerts the person to the onset of hypoglycaemia.
Hypoglycaemia may be prolonged by non-selective β-adrenoceptor antagonists, which may be a problem in people with diabetes mellitus who are treated with insulin (Ch. 40). Gluconeogenesis, a component of the metabolic response to hypoglycaemia, is dependent upon β2-adrenoceptor stimulation in the liver. Beta-adrenoceptor antagonists also blunt the autonomic response that alerts the person to the onset of hypoglycaemia. Central nervous system effects. These include sleep disturbance, vivid dreams and hallucinations, and are more common with lipophilic drugs, which readily cross the blood–brain barrier. Other consequences of CNS action include fatigue and subtle psychomotor effects, for example lack of concentration and sexual dysfunction.
Central nervous system effects. These include sleep disturbance, vivid dreams and hallucinations, and are more common with lipophilic drugs, which readily cross the blood–brain barrier. Other consequences of CNS action include fatigue and subtle psychomotor effects, for example lack of concentration and sexual dysfunction. Effects on blood lipid levels. Most β-adrenoceptor antagonists raise the plasma concentration of triglycerides and lower the concentration of high-density lipoprotein (HDL) cholesterol (Ch. 48). These changes are modest but potentially atherogenic. They are most marked with non-selective β-adrenoceptor antagonists, and least if the drug has partial agonist activity.
Effects on blood lipid levels. Most β-adrenoceptor antagonists raise the plasma concentration of triglycerides and lower the concentration of high-density lipoprotein (HDL) cholesterol (Ch. 48). These changes are modest but potentially atherogenic. They are most marked with non-selective β-adrenoceptor antagonists, and least if the drug has partial agonist activity. Sudden withdrawal syndrome. Upregulation of β-adrenoceptors (Ch. 1) during long-term treatment makes the heart more sensitive to catecholamines. Palpitation due to a greater awareness of the heart is common on withdrawal. Beta-adrenoceptor antagonists should be stopped gradually in people with ischaemic heart disease, to avoid precipitating unstable angina or myocardial infarction.
Sudden withdrawal syndrome. Upregulation of β-adrenoceptors (Ch. 1) during long-term treatment makes the heart more sensitive to catecholamines. Palpitation due to a greater awareness of the heart is common on withdrawal. Beta-adrenoceptor antagonists should be stopped gradually in people with ischaemic heart disease, to avoid precipitating unstable angina or myocardial infarction.
Calcium channel blockers
Mechanism of action and effects
Calcium is essential for excitation/contraction coupling in muscle cells. The following mechanisms of regulating intracellular free Ca2+ concentration are important pharmacologically (Figs 5.4 and 5.5).
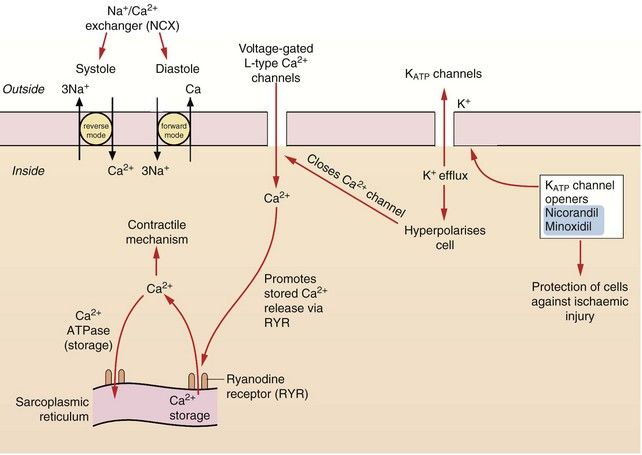
Fig. 5.4 The control of calcium regulation and actions of potassium channel openers in cardiac myocytes and blood vessels.
Calcium concentrations in cardiac cells and in vascular smooth muscle are under the control of a number of different mechanisms. Calcium entry through voltage-gated L-type Ca2+ channels stimulates ryanodine receptors (RYR) in the sarcoplasmic reticulum, releasing stored Ca2+ (known as Ca2+-induced calcium release, CICR). Intracellular Ca2+ is also regulated by exchange with Na+ via the Na+/Ca2+ exchangers (NCX) in the cell membrane. Vascular smooth muscle cells have ATP-sensitive inward rectifier K+ channels (KIR) which combine with sulfonylurea receptors to form ATP-sensitive K+ channels (KATP). Hyperpolarisation of the cell by drugs which open KATP channels, such as nicorandil, closes voltage-gated L-type Ca2+ channels and causes relaxation.
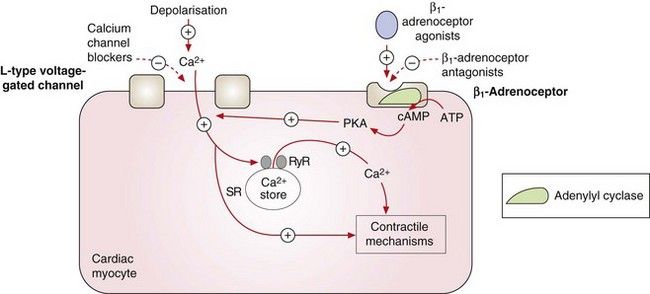
Fig. 5.5 Contraction of the cardiac myocyte by voltage-gated and receptor-operated channels.
Depolarisation during the action potential activates the voltage-gated L-type Ca2+ channels and the influx of Ca2+ into the cell results in myosin phosphorylation and muscle contraction. It also promotes further Ca2+ release from the sarcoplasmic reticulum (SR) by stimulation of ryanodine receptors (RyR). Stimulation of the β1-adrenoceptors by catecholamines activates adenylyl cyclase and the generated cAMP binds to subunits of protein kinase A (PKA), which phosphorylates the L-type Ca2+ channels, increasing their opening time and facilitating Ca2+ entry. The L-type Ca2+ channels can also be activated by other pathways, such as phospholipase C-dependent signalling triggered by agonism of α1-adrenoceptors (not shown). The activity of the voltage-gated L-type Ca2+ channels can therefore be reduced directly by calcium channel blockers or indirectly by antagonists of β1-adrenoceptors or other receptors. +, Stimulates activity; –, inhibits activity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


