Figure 3-1. Ligand-gated ion channel gatekeeper. This schematic shows a ligand-gated ion channel. In panel A, a receptor is serving as a molecular gatekeeper that acts on instruction from neurotransmission to open the channel and allow ions to travel into the cell. In panel B, the gatekeeper is keeping the channel closed so that ions cannot get into the cell. Ligand-gated ion channels are a type of receptor that forms an ion channel and are thus also called ion-channel-linked receptors or ionotropic receptors.
Numerous drugs act at many sites around such receptor/ion-channel complexes, leading to a wide variety of modifications of receptor/ion-channel actions. These modifications not only immediately alter the flow of ions through the channels, but with a delay can also change the downstream events that result from transduction of the signal that begins at these receptors. The downstream actions have been extensively discussed in Chapter 1 and include both activation and inactivation of phosphoproteins, shifting the activity of enzymes, the sensitivity of receptors, and the conductivity of ion channels. Other downstream actions include changes in gene expression and thus changes in which proteins are synthesized and which functions are amplified. Such functions can range from synaptogenesis, to receptor and enzyme synthesis, to communication with downstream neurons innervated by the neuron with the ionotropic receptor, and many more. The reader should have a good command of the function of signal transduction pathways described in Chapter 1 in order to understand how drugs acting at ligand-gated ion channels modify the signal transduction that arises from these receptors.
Drug-induced modifications in signal transduction from ionotropic (sometimes called ionotrophic) receptors can have profound actions on psychiatric symptoms. About a fifth of psychotropic drugs currently utilized in clinical practice, including many drugs for the treatment of anxiety and insomnia such as the benzodiazepines, are known to act at these receptors. Because ionotropic receptors immediately change the flow of ions, drugs that act on these receptors can have an almost immediate effect, which is why many anxiolytics and hypnotics that act at these receptors may have immediate clinical onset. This is in contrast to the actions of many drugs at G-protein-linked receptors described in Chapter 2, some of which have clinical effects – such as antidepressant actions – that may occur with a delay necessitated by awaiting initiation of changes in cellular functions activated through the signal transduction cascade. Here we will describe how various drugs stimulate or block various molecular sites around the receptor/ion-channel complex. Throughout the textbook we will show how specific drugs acting at specific ionotropic receptors have specific actions on specific psychiatric disorders.
Ligand-gated ion channels: structure and function
Are ligand-gated ion channels receptors or ion channels? The answer is “yes” – ligand-gated ion channels are a type of receptor and they also form an ion channel. That is why they are called not only a channel (ligand-gated ion channel) but also a receptor (ionotropic receptor or ion-channel-linked receptor). These terms try to capture the dual function of these ion channels/receptors.
Ligand-gated ion channels comprise several long strings of amino acids assembled as subunits around an ion channel. Decorating these subunits are also multiple binding sites for everything from neurotransmitters to ions to drugs. That is, these complex proteins have several sites where some ions travel through a channel and others also bind to the channel; where one neurotransmitter or even two cotransmitters act at separate and distinct binding sites; where numerous allosteric modulators – i.e., natural substances or drugs that bind to a site different than where the neurotransmitter binds – increase or decrease the sensitivity of channel opening.
Pentameric subtypes
Many ligand-gated ion channels are assembled from five protein subunits; that is why they are called pentameric. The subunits for pentameric subtypes of ligand-gated ion channels each have four transmembrane regions (Figure 3-2A). These membrane proteins go in and out of the membrane four times (Figure 3-2A). When five copies of these subunits are selected (Figure 3-2B), they come together in space to form a fully functional pentameric receptor with the ion channel in the middle (Figure 3-2C). The receptor sites are in various locations on each of the subunits; some binding sites are in the channel, but many are present at different locations outside the channel. This pentameric structure is typical for GABAA receptors, nicotinic cholinergic receptors, serotonin 5HT3 receptors, and glycine receptors (Table 3-1). Drugs that act directly on pentameric ligand-gated ion channels are listed in Table 3-2.
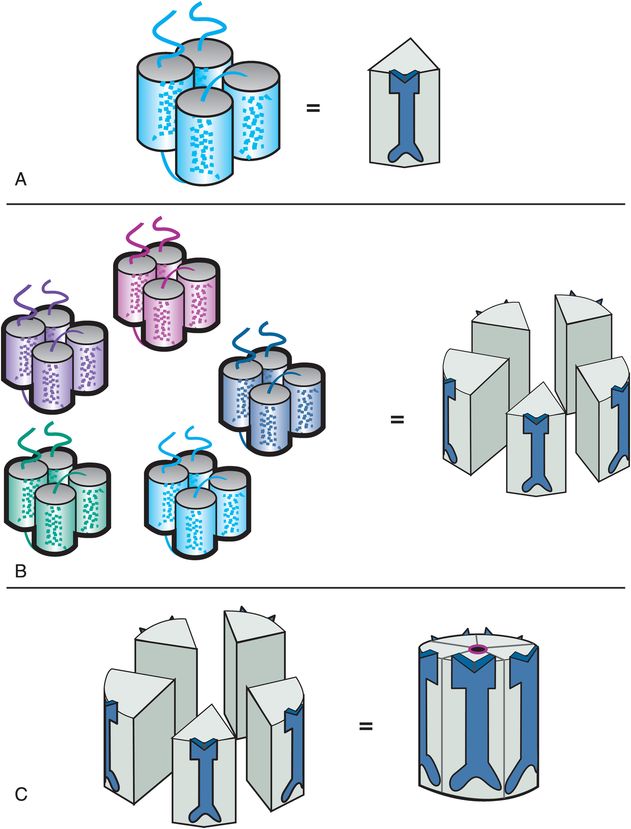
Figure 3-2. Ligand-gated ion channel structure. The four transmembrane regions of a single subunit of a pentameric ligand-gated ion channel form a cluster, as shown in panel A. An icon for this subunit is shown on the right in panel A. Five copies of the subunits come together in space (panel B) to form a functional ion channel in the middle (panel C). Pentameric ligand-gated ion channels have receptor binding sites located on all five subunits, both inside and outside the channel.
| 4 transmembrane regions 5 subunits | |
|---|---|
| Neurotransmitter | Receptor subtype |
| Acetylcholine | Nicotinic receptors (e.g., α7-nicotinic receptors; α4β2-nicotinic receptors) |
| GABA | GABAA receptors (e.g., α1 subunits) |
| Glycine | Strychnine-sensitive glycine receptors |
| Serotonin | 5HT3 receptors |
| Neurotransmitter | Ligand-gated ion-channel receptor subtype directly targeted | Pharmacologic action | Drug class | Therapeutic action |
|---|---|---|---|---|
| Acetylcholine | α4β2-nicotinic receptors | Partial agonist | Nicotinic receptor partial agonist (NRPA) (varenicline) | Smoking cessation |
| GABA | GABAA benzodiazepine receptors | Full agonist | Benzodiazepines | Anxiolytic |
| GABAA non-benzodiazepine PAM sites | Full agonist | “Z drugs”/hypnotics (zolpidem, zaleplon, zopiclone, eszopiclone) | Improve insomnia | |
| Glutamate | NMDA NAM channel sites/Mg++ sites | Antagonist | NMDA glutamate antagonist (memantine) | Slowing progression in Alzheimer’s disease |
| NMDA open channel sites | Antagonist | PCP (phencyclidine) Ketamine | Hallucinogen anesthetic | |
| Serotonin | 5HT3 | Antagonist | Antidepressant (mirtazapine) | Unknown; reduce nausea |
| 5HT3 | Antagonist | Antiemetic | Reduce chemotherapy-induced emesis |
PAM, positive allosteric modulator; NAM, negative allosteric modulator; NMDA, N-methyl-D-aspartate; Mg, magnesium.
If this structure were not complicated enough, pentameric ionotropic receptors actually have many different subtypes. Subtypes of pentameric ionotropic receptors are defined based upon which forms of each of the five subunits are chosen for assembly into a fully constituted receptor. That is, there are several subtypes for each of the four transmembrane subunits, making it possible to piece together several different constellations of fully constituted receptors. Although the natural neurotransmitter binds to every subtype of ionotropic receptor, some drugs used in clinical practice, and many more in clinical trials, are able to bind selectively to one or more of these subtypes, but not to others. This may have functional and clinical consequences. Specific receptor subtypes and the specific drugs that bind to them selectively are discussed in chapters that cover their specific clinical use.
Tetrameric subtypes
Ionotropic glutamate receptors have a different structure from the pentameric ionotropic receptors just discussed. The ligand-gated ion channels for glutamate comprise subunits that have three full transmembrane regions and a fourth re-entrant loop (Figure 3-3A), rather than four full transmembrane regions as shown in Figure 3-2A. When four copies of these subunits are selected (Figure 3-3B), they come together in space to form a fully functional ion channel in the middle with the four re-entrant loops lining the ion channel (Figure 3-3C). Thus, tetrameric subtypes of ion channels (Figure 3-3) are analogous to pentameric subtypes of ion channels (Figure 3-2), but have just four subunits rather than five. Receptor sites are in various locations on each of the subunits; some binding sites are in the channel, but many are present at different locations outside the channel.
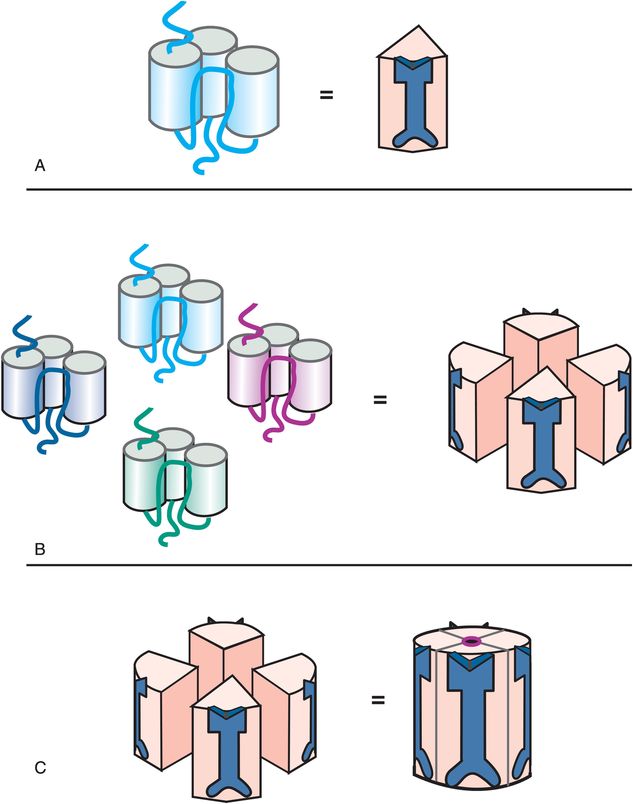
Figure 3-3. Tetrameric ligand-gated ion channel structure. A single subunit of a tetrameric ligand-gated ion channel is shown to form a cluster in panel A, with an icon for this subunit shown on the right in panel A. Four copies of these subunits come together in space (panel B) to form a functional ion channel in the middle (panel C). Tetrameric ligand-gated ion channels have receptor binding sites located on all four subunits, both inside and outside the channel.
This tetrameric structure is typical of the ionotropic glutamate receptors known as AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid) and NMDA (N-methyl-D-aspartate) subtypes (Table 3-3). Drugs that act directly at tetrameric ionotropic glutamate receptors are listed in Table 3-2. Receptor subtypes for glutamate according to the selective agonist acting at that receptor, as well as the specific molecular subunits that comprise that subtype, are listed in Table 3-3. Subtype-selective drugs for ionotropic glutamate receptors are under investigation but not currently used in clinical practice.
| 3 transmembrane regions and one re-entrant loop 4 subunits | |
|---|---|
| Neurotransmitter | Receptor subtype |
| Glutamate | AMPA (e.g., GluR1–4 subunits) |
| KAINATE (e.g., GluR5–7, KA1–2 subunits) | |
| NMDA (e.g., NMDAR1, NMDAR2A–D, NMDAR3A subunits) | |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid; NMDA, N-methyl-D-aspartate.
The agonist spectrum
The concept of an agonist spectrum for G-protein-linked receptors, discussed extensively in Chapter 2, can also be applied to ligand-gated ion channels (Figure 3-4). Thus, full agonists change the conformation of the receptor to open the ion channel the maximal amount and frequency allowed by that binding site (Figure 3-5). This then triggers the maximal amount of downstream signal transduction possible to be mediated by this binding site. The ion channel can open to an even greater extent (i.e., more frequently) than with a full agonist alone, but this requires the help of a second receptor site, that of a positive allosteric modulator, or PAM, as will be shown later.
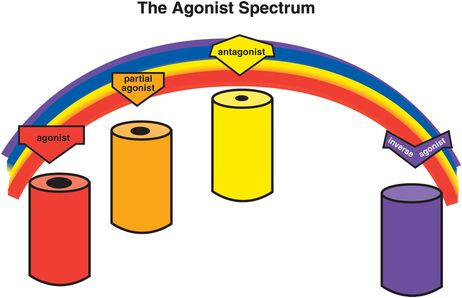
Figure 3-4. Agonist spectrum. The agonist spectrum and its corresponding effects on the ion channel are shown here. This spectrum ranges from agonists (on the far left), which open the channel the maximal amount and frequency allowed by that binding site, through antagonists (middle of the spectrum), which retain the resting state with infrequent opening of the channel, to inverse agonists (on the far right), which put the ion channel into a closed and inactive state. Between agonists and antagonists are partial agonists, which increase the degree and frequency of ion-channel opening as compared to the resting state, but not as much as a full agonist. Antagonists can block anything in the agonist spectrum, returning the ion channel to the resting state in each instance.
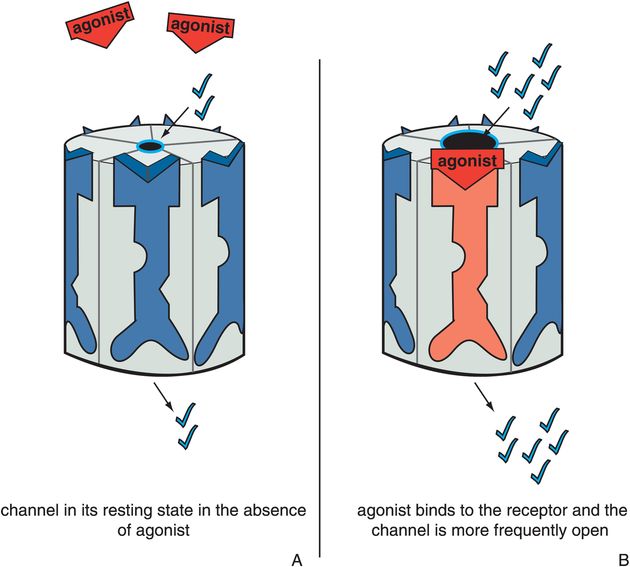
Figure 3-5. Actions of an agonist. In panel A, the ion channel is in its resting state, during which the channel opens infrequently (constitutive activity). In panel B, the agonist occupies its binding site on the ligand-gated ion channel, increasing the frequency at which the channel opens. This is represented as the red agonist turning the receptor red and opening the ion channel.
Antagonists stabilize the receptor in the resting state, which is the same as the state of the receptor in the absence of agonist (Figure 3-6). Since there is no difference between the presence and absence of the antagonist, the antagonist is said to be neutral or silent. The resting state is not a fully closed ion channel, so there is some degree of ion flow through the channel even in the absence of agonist (Figure 3-6A) and even in the presence of antagonist (Figure 3-6B). This is due to occasional and infrequent opening of the channel even when an agonist is not present and even when an antagonist is present. This is called constitutive activity and is also discussed in Chapter 2 for G-protein-linked receptors. Antagonists of ion-channel-linked receptors reverse the action of agonists (Figure 3-7) and bring the receptor conformation back to the resting baseline state, but do not block any constitutive activity.
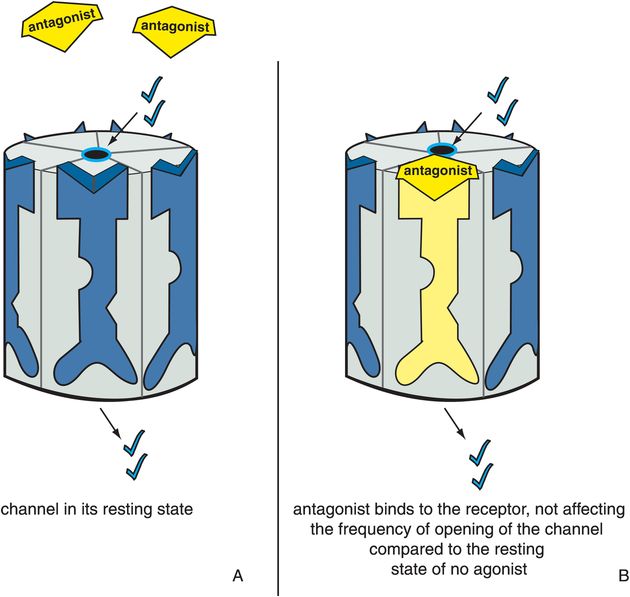
Figure 3-6. Antagonists acting alone. In panel A, the ion channel is in its resting state, during which the channel opens infrequently. In panel B, the antagonist occupies the binding site normally occupied by the agonist on the ligand-gated ion channel. However, there is no consequence to this, and the ion channel does not affect the degree or frequency of opening of the channel compared to the resting state. This is represented as the yellow antagonist docking into the binding site and turning the receptor yellow but not affecting the state of the ion channel.
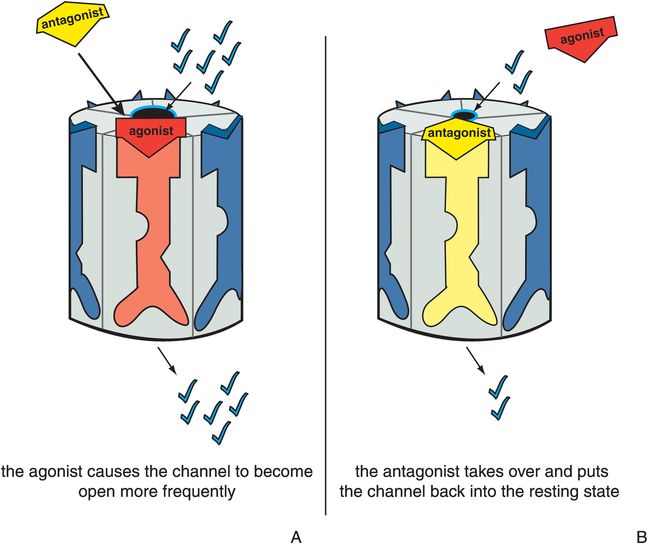
Figure 3-7. Antagonist acting in presence of agonist. In panel A, the ion channel is bound by an agonist, which causes it to open at a greater frequency than in the resting state. This is represented as the red agonist turning the receptor red and opening the ion channel as it docks into its binding site. In panel B, the yellow antagonist prevails and shoves the red agonist off the binding site, reversing the agonist’s actions and restoring the resting state. Thus, the ion channel has returned to its status before the agonist acted.
Partial agonists produce a change in receptor conformation such that the ion channel opens to a greater extent and more frequently than in its resting state but less than in the presence of a full agonist (Figures 3-8 and 3-9). An antagonist reverses a partial agonist, just as it reverses a full agonist, returning the receptor to its resting state (Figure 3-10). Partial agonists thus produce ion flow and downstream signal transduction that is something more than the resting state in the absence of agonist, yet something less than a full agonist. Just as is the case for G-protein-linked receptors, how close this partial agonist is to a full agonist or to a silent antagonist on the agonist spectrum will determine the impact of a partial agonist on downstream signal transduction events.
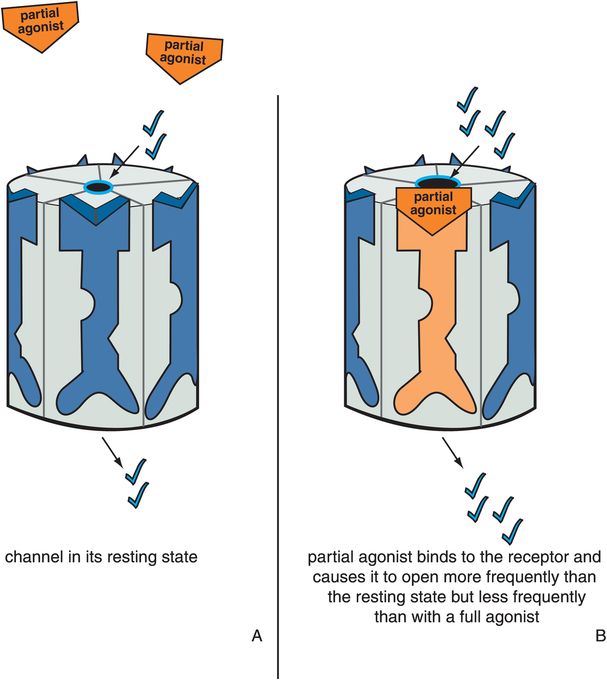
Figure 3-8. Actions of a partial agonist. In panel A, the ion channel is in its resting state and opens infrequently. In panel B, the partial agonist occupies its binding site on the ligand-gated ion channel and produces a conformational change such that the ion channel opens to a greater extent and at a greater frequency than in the resting state, though less than in the presence of a full agonist. This is depicted by the orange partial agonist turning the receptor orange and partially but not fully opening the ion channel.
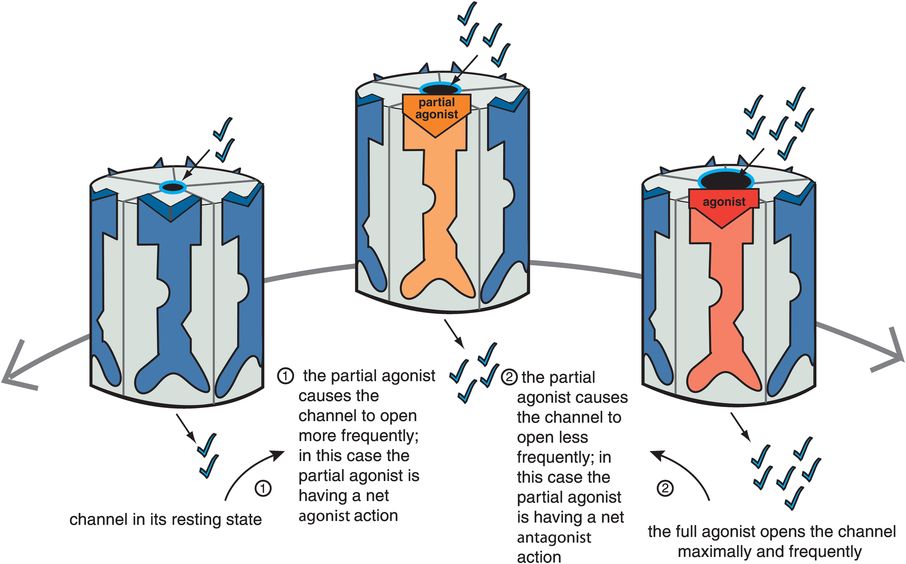
Figure 3-9. Net effect of partial agonist. Partial agonists act either as net agonists or as net antagonists, depending on the amount of agonist present. When full agonist is absent (on the far left), a partial agonist causes the channel to open more frequently as compared to the resting state and thus has a net agonist action (moving from left to right). However, in the presence of a full agonist (on the far right), a partial agonist decreases the frequency of channel opening in comparison to the full agonist and thus acts as a net antagonist (moving from right to left).
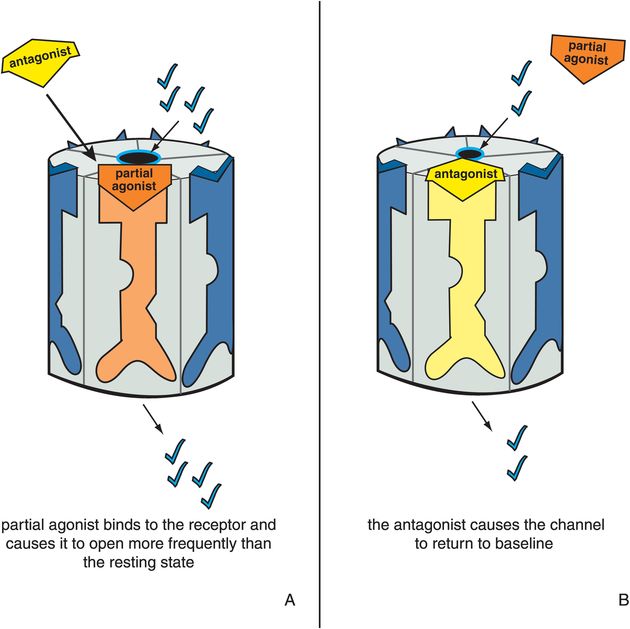
Figure 3-10. Antagonist acting in presence of partial agonist. In panel A, a partial agonist occupies its binding site and causes the ion channel to open more frequently than in the resting state. This is represented as the orange partial agonist docking to its binding site, turning the receptor orange, and partially opening the ion channel. In panel B, the yellow antagonist prevails and shoves the orange partial agonist off the binding site, reversing the partial agonist’s actions. Thus the ion channel is returned to its resting state.
The ideal therapeutic agent in some cases may need to have ion flow and signal transduction that is not too hot, yet not too cold, but just right, called the “Goldilocks” solution in Chapter 2, a concept that can apply here to ligand-gated ion channels as well. Such an ideal state may vary from one clinical situation to another, depending upon the balance between full agonism and silent antagonism that is desired. In cases where there is unstable neurotransmission throughout the brain, finding such a balance may stabilize receptor output somewhere between too much and too little downstream action. For this reason, partial agonists are also called “stabilizers,” since they have the theoretical capacity to find the stable solution between the extremes of too much full agonist action and no agonist action at all (Figure 3-9).
Just as is the case for G-protein-linked receptors, partial agonists at ligand-gated ion channels can appear as net agonists, or as net antagonists, depending upon the amount of naturally occurring full agonist neurotransmitter that is present. Thus, when a full agonist neurotransmitter is absent, a partial agonist will be a net agonist (Figure 3-9). That is, from the resting state, a partial agonist initiates somewhat of an increase in the ion flow and downstream signal transduction cascade from the ion-channel-linked receptor. However, when full agonist neurotransmitter is present, the same partial agonist will become a net antagonist (Figure 3-9): it will decrease the level of full signal output to a lesser level, but not to zero. Thus, a partial agonist can simultaneously boost deficient neurotransmitter activity yet block excessive neurotransmitter activity, another reason that partial agonists are called stabilizers. An agonist and an antagonist in the same molecule acting at ligand-gated ion channels is quite an interesting new dimension to therapeutics. This concept has led to proposals that partial agonists could treat not only states that are theoretically deficient in full agonist, but also states that are theoretically in excess of full agonist. As mentioned in the discussion of G-protein-linked receptors in Chapter 2, a partial agonist at ligand-gated ion channels could also theoretically treat states that are mixtures of both excessive and deficient neurotransmitter activity. Partial agonists at ligand-gated ion channels are just beginning to enter use in clinical practice (Table 3-2), and several more are in clinical development.
Inverse agonists at ligand-gated ion channels are different from simple antagonists, and are neither neutral nor silent. Inverse agonists are explained in Chapter 2 in relation to G-protein-linked receptors. Inverse agonists at ligand-gated ion channels are thought to produce a conformational change in these receptors that first closes the channel and then stabilizes it in an inactive form (Figure 3-11). Thus, this inactive conformation (Figure 3-11B) produces a functional reduction in ion flow and in consequent signal transduction compared to the resting state (Figure 3-11A) that is even less than that produced when there is either no agonist present or when a silent antagonist is present. Antagonists reverse this inactive state caused by inverse agonists, returning the channel to the resting state (Figure 3-12).
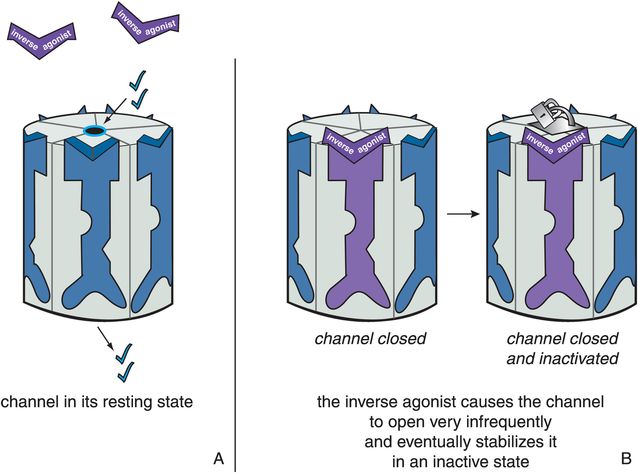
Figure 3-11. Actions of an inverse agonist. In panel A, the ion channel is in its resting state and opens infrequently. In panel B, the inverse agonist occupies the binding site on the ligand-gated ion channel and causes it to close. This is the opposite of what an agonist does and is represented by the purple inverse agonist turning the receptor purple and closing the ion channel. Eventually, the inverse agonist stabilizes the ion channel in an inactive state, represented by the padlock on the channel itself.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


