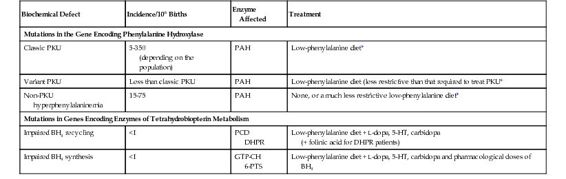
Phenylketonuria.
Classic PKU is the epitome of the enzymopathies. It results from mutations in the gene encoding PAH, which converts phenylalanine to tyrosine (see Fig. 12-2 and Table 12-1). The discovery of PKU in 1934 marked the first demonstration of a genetic defect as a cause of intellectual disability. Because patients with PKU cannot degrade phenylalanine, it accumulates in body fluids and damages the developing central nervous system in early childhood. A small fraction of phenylalanine is metabolized to produce increased amounts of phenylpyruvic acid, the keto acid responsible for the name of the disease. Ironically, although the enzymatic defect has been known for many decades, the precise pathogenetic mechanism(s) by which increased phenylalanine damages the brain is still uncertain. Importantly, the neurological damage is largely avoided by reducing the dietary intake of phenylalanine. The management of PKU is a paradigm of the treatment of the many metabolic diseases whose outcome can be improved by preventing accumulation of an enzyme substrate and its derivatives; this therapeutic principle is described further in Chapter 13.
Variant Phenylketonuria and Nonphenylketonuria Hyperphenylalaninemia.
Whereas PKU results from a virtual absence of PAH activity (less than 1% of that in controls), less severe phenotypes, designated non-PKU hyperphenylalaninemia and variant PKU (see Table 12-1), result when the mutant PAH enzyme has some residual activity. The fact that a very small amount of residual enzyme activity can have a large impact on phenotype is another general principle of the enzymopathies (see Box).
Mutant Enzymes and Disease
General Concepts
The following concepts are fundamental to the understanding and treatment of enzymopathies.
• Inheritance patterns
Enzymopathies are almost always recessive or X-linked (see Chapter 7). Most enzymes are produced in quantities significantly in excess of minimal biochemical requirements, so that heterozygotes (typically with approximately 50% of residual activity) are clinically normal. In fact, many enzymes may maintain normal substrate and product levels with activities of less than 10%, a point relevant to the design of therapeutic strategies (e.g., homocystinuria due to cystathionine synthase deficiency—see Chapter 13). The enzymes of porphyrin synthesis are exceptions (see discussion of acute intermittent porphyria in main text, later).
• Substrate accumulation or product deficiency
Because the function of an enzyme is to convert a substrate to a product, all of the pathophysiological consequences of enzymopathies can be attributed to the accumulation of the substrate (as in PKU), to the deficiency of the product (as in glucose-6-phosphate dehydrogenase deficiency (Case 19), or to some combination of the two (Fig. 12-3).
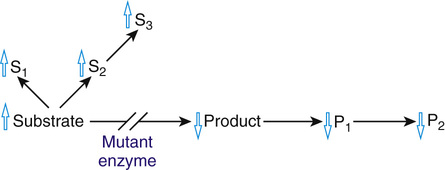
• Diffusible versus macromolecular substrates
An important distinction can be made between enzyme defects in which the substrate is a small molecule (such as phenylalanine, which can be readily distributed throughout body fluids by diffusion or transport) and defects in which the substrate is a macromolecule (such as a mucopolysaccharide, which remains trapped within its organelle or cell). The pathological change of the macromolecular diseases, such as Tay-Sachs disease, is confined to the tissues in which the substrate accumulates. In contrast, the site of the disease in the small molecule disorders is often unpredictable, because the unmetabolized substrate or its derivatives can move freely throughout the body, damaging cells that may normally have no relationship to the affected enzyme, as in PKU.
• Loss of multiple enzyme activities
A patient with a single-gene defect may have a loss of function in more than one enzyme. There are several possible mechanisms: the enzymes may use the same cofactor (e.g., BH4 deficiency); the enzymes may share a common subunit or an activating, processing, or stabilizing protein (e.g., the GM2 gangliosidoses); the enzymes may all be processed by a common modifying enzyme, and in its absence, they may be inactive, or their uptake into an organelle may be impaired (e.g., I-cell disease, in which failure to add mannose 6-phosphate to many lysosomal enzymes abrogates the ability of cells to recognize and import the enzymes); and a group of enzymes may be absent or ineffective if the organelle in which they are normally found is not formed or is abnormal (e.g., Zellweger syndrome, a disorder of peroxisome biogenesis).
• Phenotypic homology
The pathological and clinical features resulting from an enzyme defect are often shared by diseases due to deficiencies of other enzymes that function in the same area of metabolism (e.g., the mucopolysaccharidoses) as well as by the different phenotypes that can result from partial versus complete defects of one enzyme. Partial defects often present with clinical abnormalities that are a subset of those found with the complete deficiency, although the etiological relationship between the two diseases may not be immediately obvious. For example, partial deficiency of the purine enzyme hypoxanthine-guanine phosphoribosyltransferase causes only hyperuricemia, whereas a complete deficiency causes hyperuricemia as well as a profound neurological disease, Lesch-Nyhan syndrome, which resembles cerebral palsy.
Variant PKU includes patients who require only some dietary phenylalanine restriction but to a lesser degree than that required in classic PKU, because their increases in blood phenylalanine levels are more moderate and less damaging to the brain. In contrast to classic PKU, in which the plasma phenylalanine levels are greater than 1000 μmol/L when the patient is receiving a normal diet, non-PKU hyperphenylalaninemia is defined by plasma phenylalanine concentrations above the upper limit of normal (120 μmol/L), but less than the levels seen in classic PKU. If the increase in non-PKU hyperphenylalaninemia is small (<400 μmol/L), no treatment is required; these individuals come to clinical attention only because they are identified by newborn screening (see Chapter 17). Their normal phenotype has been the best indication of the “safe” level of plasma phenylalanine that must not be exceeded in treating classic PKU. The association of these three clinical phenotypes with mutations in the PAH gene is a clear example of allelic heterogeneity leading to clinical heterogeneity (see Table 12-1).
Allelic and Locus Heterogeneity in the Hyperphenylalaninemias
Allelic Heterogeneity in the PAH Gene.
A striking degree of allelic heterogeneity at the PAH locus—more than 700 different mutations worldwide—has been identified in patients with hyperphenylalaninemia associated with classic PKU, variant PKU, and non-PKU hyperphenylalaninemia (see Table 12-1). Seven mutations account for a majority of known mutant alleles in populations of European descent, whereas six others represent the majority of PAH mutations in Asian populations (Fig. 12-4). The remaining disease-causing mutations are individually rare. To record and make this information publicly available, a PAH database has been developed by an international consortium.
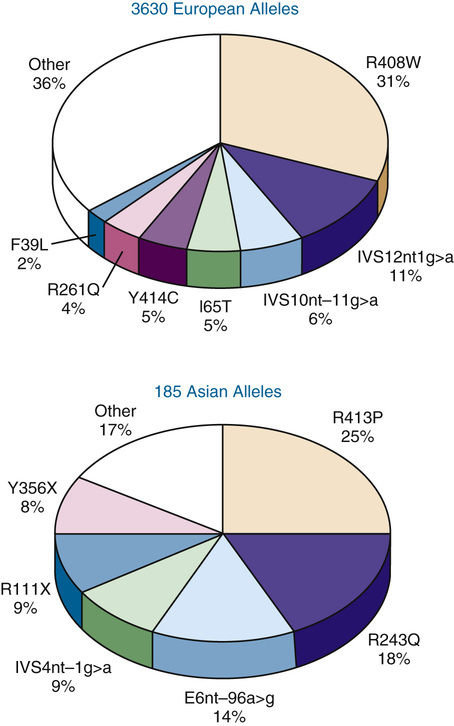
The allelic heterogeneity at the PAH locus has major clinical consequences. Most important is the fact that most hyperphenylalaninemic subjects are compound heterozygotes (i.e., they have two different disease-causing alleles) (see Chapter 7). This allelic heterogeneity accounts for much of the enzymatic and phenotypic heterogeneity observed in this patient population. Thus, mutations that eliminate or dramatically reduce PAH activity generally cause classic PKU, whereas greater residual enzyme activity is associated with milder phenotypes. However, homozygous patients with certain PAH mutations have been found to have phenotypes ranging all the way from classic PKU to non-PKU hyperphenylalaninemia. Accordingly, it is now clear that other unidentified biological variables—undoubtedly including modifier genes—generate variation in the phenotype seen with any specific genotype. This lack of a strict genotype-phenotype correlation, initially somewhat surprising, is now recognized to be a common feature of many single-gene diseases and highlights the fact that even monogenic traits like PKU are not genetically “simple” disorders.
Defects in Tetrahydrobiopterin Metabolism.
In 1% to 3% of hyperphenylalaninemic patients, the PAH gene is normal, and the hyperphenylalaninemia results from a defect in one of the steps in the biosynthesis or regeneration of BH4, the cofactor for PAH (see Table 12-1 and Fig. 12-2). The association of a single biochemical phenotype, such as hyperphenylalaninemia, with mutations in different genes, is an example of locus heterogeneity (see Table 11-1). The proteins encoded by genes that manifest locus heterogeneity generally act at different steps in a single biochemical pathway, another principle of genetic disease illustrated by the genes associated with hyperphenylalaninemia (see Fig. 12-2). BH4-deficient patients were first recognized because they developed profound neurological problems in early life, despite the successful administration of a low-phenylalanine diet. This poor outcome is due in part to the requirement for the BH4 cofactor of two other enzymes, tyrosine hydroxylase and tryptophan hydroxylase. These hydroxylases are critical for the synthesis of the monoamine neurotransmitters dopamine, norepinephrine, epinephrine, and serotonin (see Fig. 12-2).
The locus heterogeneity of hyperphenylalaninemia is of great significance because the treatment of patients with a defect in BH4 metabolism differs markedly from subjects with mutations in PAH, in two ways. First, because the PAH enzyme of individuals with BH4 defects is itself normal, its activity can be restored by large doses of oral BH4, leading to a reduction in their plasma phenylalanine levels. This practice highlights the principle of product replacement in the treatment of some genetic disorders (see Chapter 13). Consequently, phenylalanine restriction can be significantly relaxed in the diet of patients with defects in BH4 metabolism, and some patients actually tolerate a normal (i.e., a phenylalanine-unrestricted) diet. Second, one must also try to normalize the neurotransmitters in the brains of these patients by administering the products of tyrosine hydroxylase and tryptophan hydroxylase, L-dopa and 5-hydroxytryptophan, respectively (see Fig. 12-2 and Table 12-1).
Remarkably, mutations in sepiapterin reductase, an enzyme in the BH4 synthesis pathway, do not cause hyperphenylalaninemia. In this case, only dopa-responsive dystonia is seen, due to impaired synthesis of dopamine and serotonin (see Fig. 12-2). It is thought that alternative pathways exist for the final step in BH4 synthesis, bypassing the sepiapterin reductase deficiency in peripheral tissues, an example of genetic redundancy.
For these reasons, all hyperphenylalaninemic infants must be screened to determine whether their hyperphenylalaninemia is the result of an abnormality in PAH or in BH4 metabolism. The hyperphenylalaninemias thus illustrate the critical importance of obtaining a specific molecular diagnosis in all patients with a genetic disease phenotype—the underlying genetic defect may not be what one first suspects, and the treatment can vary accordingly.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


