The term CAPA is used to define the program in quality manuals as well as in the standard operating procedures (SOPs) defining the quality system. Whether there is a single procedure that defines the process for CAPAs or whether there are separate procedures for CAPA is not important. What is important is that the process be clearly defined, that it is structured, and most critically, that the investigations have the objective of determining the root cause of the issue.
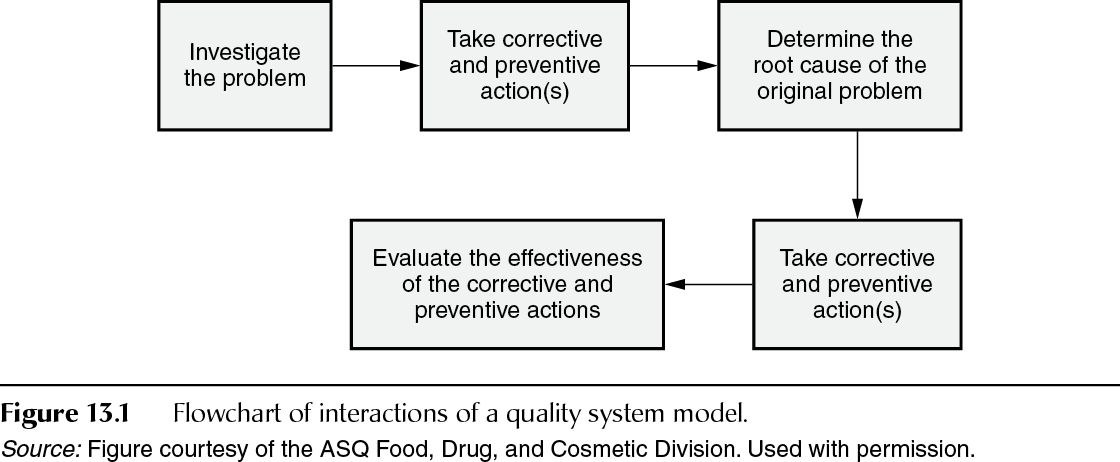
FDA guidance Quality Systems Approach to Pharmaceutical CGMP Regulations states that there are three separate concepts used in the quality system model for CAPA (Figure 13.1): remedial corrective actions to correct the immediate problem identified, root cause analysis to determine and understand the actual cause of the deviation or issue and potentially provide guidance for the prevention of a future occurrence of a similar problem or issue, and preventive action, which includes those activities carried out as a result of the root cause analysis to prevent a recurrence of the same or similar issue.
Good manufacturing practices (GMP) requirements dictate that all of the manufacturing, packaging, testing, and holding of drug product must take place according to established and approved procedures. Once the situation is such that this condition is not met, it is necessary to investigate to determine the corrective action necessary. The level of investigation, the corrective action necessary, the formality with which it is documented, and the level of control extended should be based on the risks involved (in accordance with ICH Q9). Many firms have established “alert” and “action” limits on many of their processes to assist in defining the level of formality needed. Regardless of level of formality involved, specific needs for every investigation include:
• The individuals involved in the investigation, and the decision-making authority, must have sufficient education, experience, and knowledge to carry out their associated tasks, which usually means cross-functional multidisciplinary teams including Quality Assurance.
• The investigation must be thorough, timely, unbiased, well-documented, and scientifically sound.
• It must include sufficient information to stand on its own and allow an independent reviewer (possibly a regulator) to understand the logic, the rationale for decisions, and who the decision makers were.
• It must identify the appropriate corrective action, and to properly take preventive measures, it must identify the root cause of the problem.
An investigation is a process. While the individual situation and immediate players involved may change, the steps taken, the tools employed, and the key decision makers should be well defined and not decided ad hoc.
The initial investigation usually will not determine the root cause, but will usually provide guidance on immediate corrective actions necessary to prevent further nonconformity. The investigation is not over when the initial corrective action is taken; it must continue further to determine the root cause(s). Determining root cause can be arrived at by various methods: flowcharts, fishbone diagram, process mapping, checklists, fault tree analysis, and so on. FDA prefers, whenever feasible, that statistical and qualitative tools be used to find the most likely root cause(s) by identifying trends and patterns. Whichever method is used, the rationale and the data to support the decision for the preventive action(s) should be documented and should be approved by the proper authority (as defined by company policy). Preventive measures are taken to prevent nonconformance. They are meant to prevent recurrence, even if that recurrence is at another company location; preventive measures may not always be site specific.
Who is responsible for carrying out a corrective action and who must approve the activity are usually defined in a firm’s policies and procedures. Because the need for corrective action must be determined taking into consideration the possible consequence or result of the action, the selection of who will carry out the action(s) should be based on the expertise required, the immediacy of need, and the level of risk to the product. The corrective actions taken must be documented (21 CFR 211.192) and are subject to review and approval by the quality unit. All corrective actions are expected to take place in a timely manner to prevent loss of data and loss of product integrity. Corrective actions that prompt change should go through the company’s change management system.
Once the corrective action is complete, the preventive action is implemented; there should be a plan to follow up at a specified time to ensure that the CAPAs were effective. Once the results of CAPA have been reviewed and determined to have been effective, the evaluation should be documented.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


