1 Introduction to pharmacology
After reading this chapter, you will:
Molecular basis of pharmacology
What is pharmacology?
Pharmacology is the study of the actions, mechanisms, uses and adverse effects of drugs.
Although drugs are intended to have a selective action, this is rarely achieved.
Drug names and classification
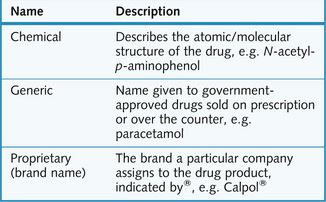
A single drug can have a variety of names (Fig. 1.1) and belong to many classes. Drugs are classified according to their:
Transport systems
Receptors
The following are naturally occurring ligands:
There are four main types of receptor (Fig. 1.2).
1 Receptors directly linked to ion channels
Receptors that are directly linked to ion channels (Fig. 1.3) are mainly involved in fast synaptic neurotransmission. A classic example of a receptor linked directly to an ion channel is the nicotinic acetylcholine receptor (nicAChR).
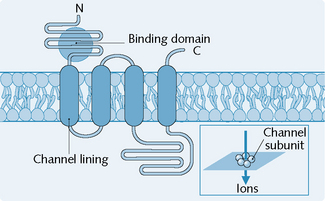
The nicAChRs possess several characteristics:
2 G-protein linked receptors
G-protein linked receptors (Fig. 1.4) are involved in relatively fast transduction. G-protein linked receptors are the predomínant receptor type in the body; muscarinic, ACh, adrenergic, dopamine, serotonin and opiate receptors are all examples of G-protein linked receptors.
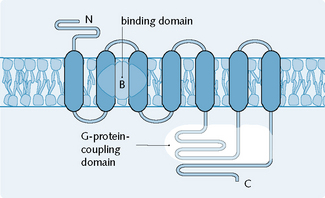
Fig. 1.4 General structure of the subunits of receptors linked to G-proteins. (C, C-terminal; N, N-terminal.)
(Redrawn from Page et al. 2006.)
G-proteins
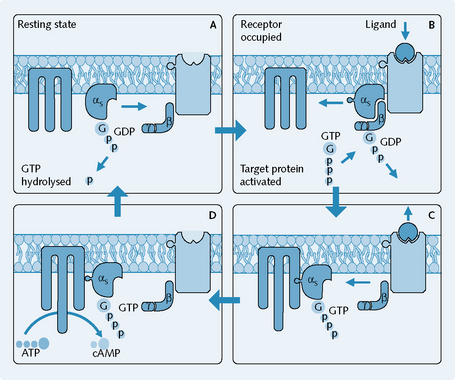
Figure 1.5 illustrates the mechanism of G-protein linked receptors:
Targets for G-proteins
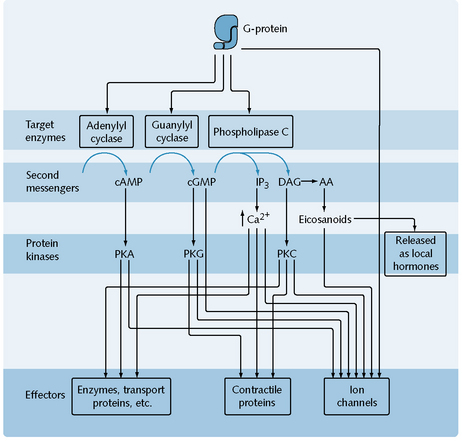
G-proteins interact with either ion channels or secondary messengers. G-proteins may activate ion channels directly, e.g. muscarinic receptors in the heart are linked to potassium channels which open directly on interaction with the G-protein, causing a slowing down of the heart rate. Secondary messengers are a family of mediating chemicals that transduces the receptor activation in to a cellular response. These mediators can be targeted and three main secondary messenger systems exist as targets of G-proteins (Fig. 1.6).
Drug–receptor interactions
Most drugs produce their effects by acting through specific protein molecules called receptors.
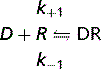
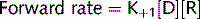
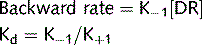
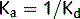
Agonists
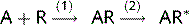
where: (1) affinity; (2) efficacy.
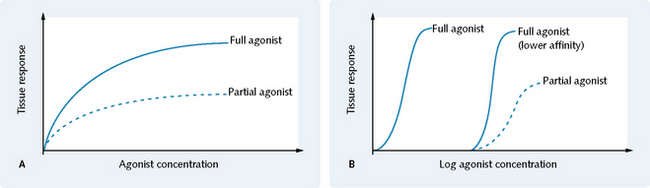
Partial agonists cannot bring about the same maximum response as full agonists, even if their affinity for the receptor is the same (Fig. 1.7).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


