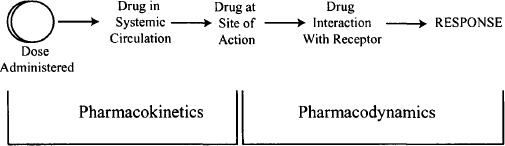
The steps between drug input and the emergence of the response can be broken down into two phases: pharmacokinetic and pharmacodynamic. The pharmacokinetic phase encompasses all the events between the administration of a dose and the achievement of drug concentrations throughout the body. The pharmacodynamic phase encompasses all the events between the arrival of the drug at its site of action and the onset, magnitude, and duration of the biological response (Figure 1.1). The rational design of optimum dosing regimens must be based on a thorough understanding of these two phases, and ideally will include the development of one or more mathematical expressions for the relationship between dose and the time course of drug response.
FIGURE 1.1 The two phases of drug action. The pharmacokinetic phase is concerned with the relationship between the value of the dose administered and the value of the drug concentrations achieved in the body; the pharmacodynamic phase is concerned with the relationship between drug concentrations at the site of action and the onset, intensity, and duration of drug response.
Optimum drug administration is important not only for ensuring good patient outcomes in clinical practice but also in the design of clinical trials during drug development. The cost of drug research and development is enormous, so it is critical that all drug candidates selected for human trials be evaluated in the most efficient, cost-effective manner possible. The application of pharmacokinetic and pharmacodynamic principles to this process has been shown to enhance the selection of optimum doses and the optimum design of phase II clinical trials.
1.2 INTRODUCTION TO PHARMACODYNAMICS
Pharmaco– comes from the Greek word for “drug,” pharmackon, and dynamics means “of or relating to variation of intensity.” Pharmacodynamics (PD) is the study of the magnitude of drug response. In particular, it is the study of the onset, intensity, and duration of drug response and how these are related to the concentration of a drug at its site of action. An overview of some basic drug terminology and the drug response–concentration relationship is provided below.
1.2.1 Drug Effects at the Site of Action
Note that although some references and textbooks distinguish the terms drug effect and drug response, this distinction has not been adopted universally. In this book, effect and response are used interchangeably.
1.2.1.1 Interaction of a Drug with Its Receptor
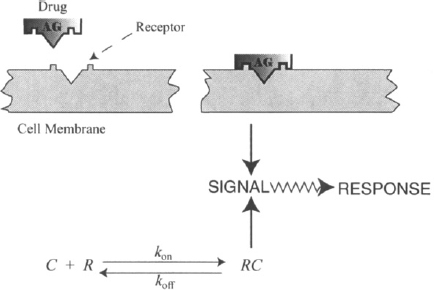
Drug response is initiated by a chemical interaction between a drug and a special binding site on a macromolecule in a tissue. This macromolecule is known as a drug receptor. The drug–receptor interaction results in a conformational change in the receptor, which results in the generation of a stimulus, which ultimately leads to a biochemical or physiological response (Figure 1.2). Most receptors (over 95%) are proteins, but other types of receptors exist. For example, the receptors of the alkylating agents used in cancer chemotherapy are on DNA. The drug–receptor interaction involves chemical bonding, which is usually reversible in nature and can be expressed using the law of mass action (Figure 1.2). Thus, at the site of action the drug binds to its receptor and equilibrium is established between the bound and the unbound drug. As the drug is eliminated from the body and removed from its site of action, it dissociates from the receptor, which is left unchanged, and the response dissipates.
FIGURE 1.2 Drug–receptor interaction. Here AG signifies a drug agonist, C is the free drug concentration (not bound to the receptor), R the concentration of free receptors, RC the concentration of the drug–receptor complex, and kon and koff are the rate constants for the forward and backward processes, respectively.
In contrast, a few drugs form irreversible covalent bonds with their receptors. For example, aspirin inhibits platelet aggregation by inhibiting the formation of thromboxane in the platelets. It accomplishes this by binding covalently to and blocking the catalytic activity of cyclooxygenase, the enzyme that produces thromboxane. The effect of a single dose of aspirin will persist long after the drug has been removed from its site of action and will continue until new cyclooxygenase molecules are synthesized, which can then resume the production of thromboxane. Other examples of drugs that bind irreversibly to their receptors include the alkylating agents mentioned above, and proton pump inhibitors such as omeprazole, which block the secretion of gastric acid by binding irreversibly to the H+,K+-ATPase pumps of parietal cells.
Owing to the chemical nature of drug–receptor interaction, it is highly dependent on the chemical structure of both the drug and the receptor; small changes in the structure of the drug can reduce or destroy activity. For example, the drug–receptor interaction can distinguish between the R- and S-isomers of drugs that have chiral carbon atoms. Usually, one isomer is much more active than the other. The S-isomer of warfarin, for example, is two to five times more active than the R-isomer. The development and promotion of S-omeprazole (Nexium) is based on the premise that the S-isomer has the higher affinity for the binding site and thus offers therapeutic advantages over preparations containing racemic mixtures (equal quantities of each isomer) of omeprazole, such as Prilosec and its generic equivalents.
Receptors are assumed to exist for all active endogenous compounds (natural ligands), such as neurotransmitters and hormones. The interaction of the natural ligands with their receptors controls and/or regulates physiological and biochemical processes in the body. In most cases, drugs mimic or antagonize the actions of endogenous ligands by interacting with their cognate receptors. For example, epinephrine is the natural ligand that interacts with the β2-adrenergic receptors in bronchial smooth muscle to bring about bronchial dilation. The drug albuterol also interacts with this receptor to produce bronchial dilation. Acetylcholine transmits signals through a synapse by interacting with its nicotinic receptor on the postsynaptic neuronal membrane. The interaction results in the production of an action potential, the response. The action of acetylcholine is mimicked by the drug nicotine.
It should be noted that there are a few drugs that do not act on receptors but that exert their action by bringing about physicochemical changes in the body. For example, conventional antacids such as calcium carbonate act as buffers to reduce acidity in the stomach, and polyethylene glycol is an osmotic laxative that acts by preventing the absorption of water in the large intestine.
1.2.1.2 Postreceptor Events
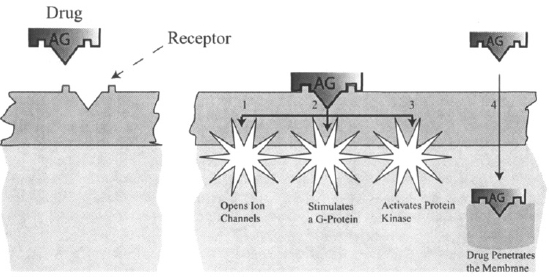
Drugs almost always bring about some type of change in the intracellular environment of cells, but the lipophilic cell membrane presents a physical barrier to most drugs and endogenous ligands. As a result, most receptors are located on the cell membrane itself. The ultimate action of a drug is dependent on the initial stimulus that results from the interaction of the drug with its membrane-bound receptor being relayed to the inside of the cell. The relaying of the initial stimulus, known as coupling or signal transduction, often involves a cascade of different steps during which the initial signal may be amplified or diminished. Some important transduction mechanisms are summarized below (see Figure 1.3).
FIGURE 1.3 Diagrammatic representations of how a drug receptor interaction brings about intracellular events. The intracellular relay of the initial signal resulting from the interaction of a drug with a membrane-bound receptor can be accomplished in one of three ways: (1) the direct opening of ion channels; (2) the activation of a G-protein that may lead to the activation of another enzyme or to a modulation of an ion channel; (3) the activation of protein kinase. Alternatively, (4), some drugs are able to penetrate membranes and directly activate intracellular receptors.
Some drugs are lipophilic enough to penetrate the cell membrane; others may be transported across the cell membrane by uptake transporters. Drugs that are able to enter a cell can interact directly with intracellular receptors. Examples include many steroids, such as glucocorticoid steroids, sex hormones, and thyroid hormones, and drugs such as the HMG-CoA reductase inhibitors (commonly known as statins), which act on enzymes within the hepatocyte, and metformin, which is Iransported across the hepatocyte by an uptake transporter.
1.2.2 Agonists, Antagonists, and Concentration–Response Relationships
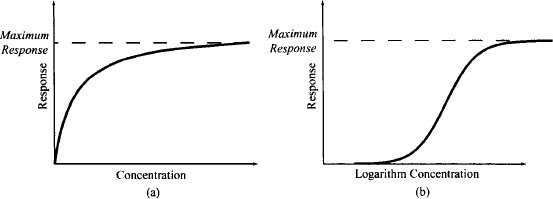
A drug that mimics the endogenous receptor ligand to activate the receptor is referred to as an agonist. The typical relationship between the drug effect and the agonist concentration at the receptor site is shown in Figure 1.4a. Note that as the concentration of the drug increases, the effect increases. At low concentrations, there is a linear relationship between concentration and effect (i.e., the response is proportional to the concentration). At higher drug concentrations, increases in concentration bring about much smaller changes in effect (the law of limited returns). Eventually, at very high concentrations, the effect achieves a maximum value and then remains constant and independent of concentration. In this area of the curve, increases in concentration will not result in further increases in response. This relationship is observed because response is generated by a saturable, capacity-limited process. For example, the response may be limited by the number of receptors that a tissue contains. At low drug concentrations there are many free receptors, so as the drug concentration increases, the drug can bind to the free receptors, and response can increase proportionally. At higher concentrations more and more of the receptors are occupied, so increases in the drug concentration produce much less increase in effect. Eventually, all of the receptors are occupied (or saturated) and a maximum effect is observed. To accommodate a wide range of concentrations, the relationship between effect and concentration is usually plotted on a semilogarithmic scale, which transforms the plot to a sigmoidal shape (Figure 1.4b).
FIGURE 1.4 Plots of response versus drug concentration: on a linear scale (a) and on a semilogarithmic scale (b).
Many agonists are able to produce the system’s maximum response without fully occupying all the receptors. In these systems the maximum response of the drug must be the result of some other saturable, capacity-limited process that occurs after receptor binding. These tissues or systems are said to have spare receptors. Experimentally, the presence of spare receptors can be demonstrated by destroying some of the receptors. If an agonist is still able to produce a maximum response, the system must contain spare receptors.
The efficiency with which a drug’s interaction with the receptor is converted into the initial stimulus or biosignal is a function of the number of receptors at the site of action and a drug’s intrinsic efficacy, which can be defined as the magnitude of the stimulus produced per unit receptor occupied. The value of the stimulus that results from a specific concentration of a drug is also a function of the drug’s affinity for its receptors. Affinity can be defined as the extent or fraction to which a drug binds to receptors at any given drug concentration. Drugs that have high affinity require less drug to produce a certain degree of binding and to elicit a certain response compared to drugs with low affinity. Affinity is one of the factors that determine potency (see Chapter 16).
A drug that binds to a receptor but does not activate it is referred to as an antagonist. The presence of an antagonist at the receptor site blocks the action of the agonist (Figure 1.5), and higher concentrations of the agonist are needed to displace the antagonist and to produce the effect that it elicited when the antagonist was absent. The antagonist shifts the concentration–response curve of an agonist to the right (Figure 1.6). At sufficiently high concentrations of the antagonist, the agonist’s action may be blocked completely and the effect of even high concentrations of the agonist reduced to zero. Some drugs bind to receptors but the binding is less efficient, and a full response cannot be achieved even when the drug’s concentration is very high and all the receptors are occupied (Figure 1.7). These drugs are referred to as partial agonists. A partial agonist will block the effect of a full agonist. In the presence of high concentrations of a partial agonist, the action of a full agonist can be reduced to the maximum response elicited by the partial agonist. Clinically, partial agonists are used to act as buffers to avoid full stimulation of a system. Examples of partial agonists include several β-blockers, including pindolol, and the opioid buprenorphine. The latter is a partial agonist on the μ-opioid receptors and is considered a safer alternative to morphine because it does not produce as much respiratory depression.
FIGURE 1.5 Diagrammatic representation of the action of an antagonist. The antagonist (ATG) binds to the receptor but does not produce a signal. Its presence on the receptor blocks the action of agonists (AG), including the natural ligand.
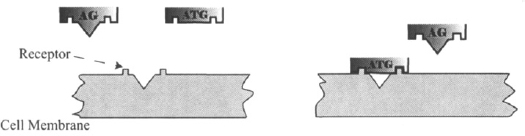
FIGURE 1.6 Plot of response versus logarithm concentration for an agonist in the absence and presence of increasing concentrations of an antagonist.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


