Chapter 17 Intracellular Messengers
A key is useless without a matching lock, and a hormone or other extracellular signaling molecule is useless without a matching receptor in its target cell. The receptor is an allosteric protein that changes its conformation when it binds the signaling molecule. Messengers that do not enter the cell activate receptors in the plasma membrane, and many of those that can enter activate receptors in the cytoplasm or nucleus (Fig. 17.1).
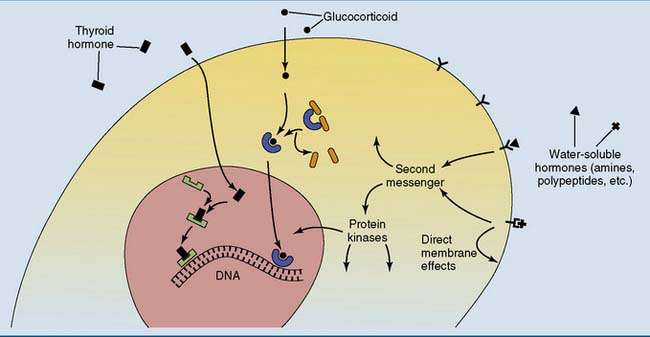
 , Thyroid hormone receptor;
, Thyroid hormone receptor;  , glucocorticoid receptor;
, glucocorticoid receptor;  and
and  , cell surface receptors.
, cell surface receptors.Receptor-hormone interactions are noncovalent, reversible, and saturable
Like the binding of a substrate to its enzyme (see Chapter 4) or an antigen to its antibody (see Chapter 15), hormone-receptor binding is always noncovalent. Being noncovalent, it is reversible. The receptor-hormone complex (RċH) can easily dissociate back into free receptor (R) and free hormone (H):
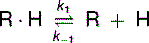
The dissociation constant KD of the receptor-hormone complex is defined as
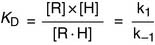
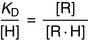
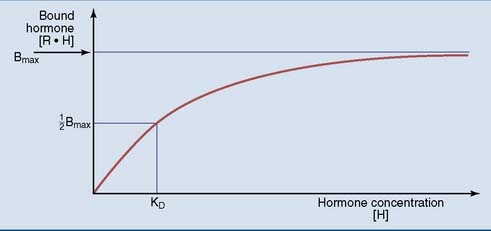
Hormone binding shows saturation kinetics (Fig. 17.2). At a hormone concentration far above KD, almost all receptors are occupied. The physiological response is near maximal and cannot be augmented by adding even more hormone. This is equivalent to zero-order kinetics for enzymes. Maximal binding (Bmax) corresponds to the number of receptor molecules in the cell.
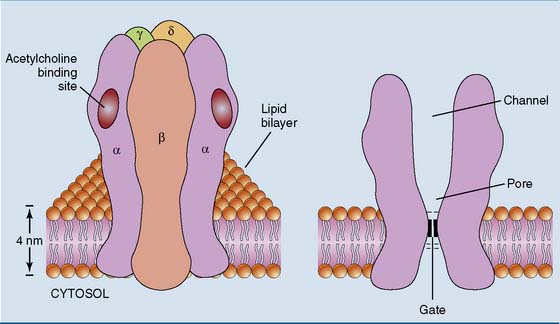
Many neurotransmitter receptors are ion channels
The nicotinic acetylcholine receptor in the neuromuscular junction is a classic example. This receptor is a channel for the monovalent cations sodium and potassium, with five subunits that each contribute to the channel (Fig. 17.3). The channel is closed in the resting state, opening only when acetylcholine binds. Opening of the channel causes a rapid influx of sodium down its electrochemical gradient, which depolarizes the membrane.
Receptors for steroid and thyroid hormones are transcription factors
An inherited deficiency of a receptor makes the cells unable to respond to the matching hormone (Clinical Examples 17.1 and 17.2).
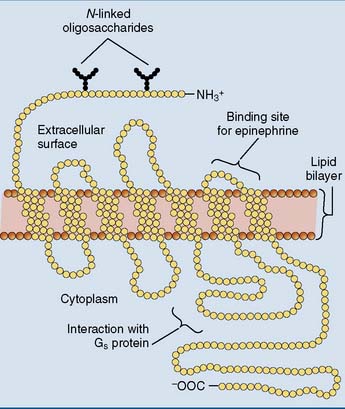
Seven-transmembrane receptors are coupled to G proteins
Most hormone receptors belong to a family of membrane proteins that crisscross the membrane seven times (Fig. 17.4). These receptors do not form a channel and possess no enzymatic activities, but they trigger their signaling cascades by activating a guanine nucleotide-binding G protein.
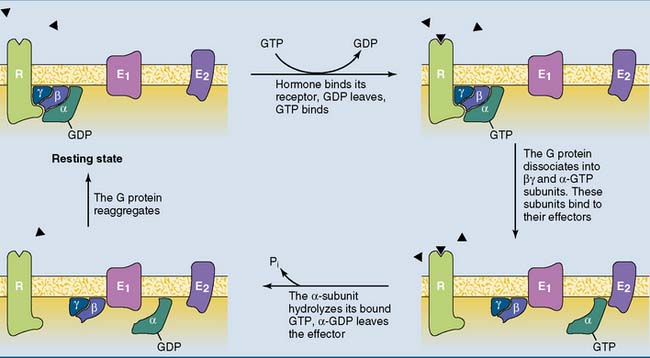
The function of the G protein is described in Figure 17.5. The inactive G protein is associated with the unstimulated receptor, with GDP bound to the α subunit. Hormone binding changes the conformation of the receptor and the attached G protein. As a result, the α subunit of the G protein loses its affinity for GDP, which dissociates away and is replaced by GTP.
Adenylate cyclase is regulated by G proteins
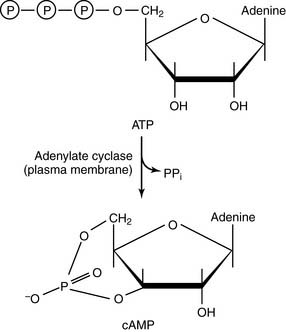
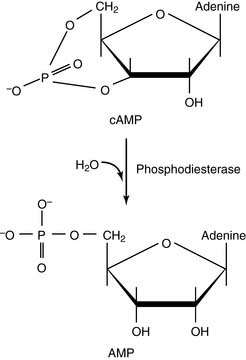
cAMP is degraded by phosphodiesterases:
CLINICAL EXAMPLE 17.3: Cholera
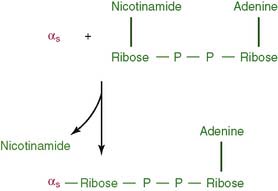
Some strains of Escherichia coli cause the common traveler’s diarrhea by raising the cAMP level with a similar toxin, although other strains do the same by raising cGMP. The best symptomatic treatment of traveler’s diarrhea is opium taken by mouth. Opiate receptors couple to the Gi protein (Table 17.1), thereby antagonizing the out-of-control Gs protein.

Hormones can both activate and inhibit the cAMP cascade
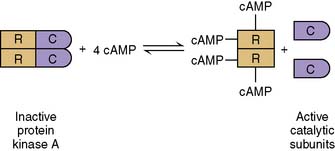
The cAMP cascade amplifies the hormonal signal (Fig. 17.6). For example, the binding of a single epinephrine molecule to a β-adrenergic receptor activates up to 20 Gs proteins. Each αs-GTP subunit activates adenylate cyclase long enough to cause the synthesis of hundreds of cAMP molecules. Although only four cAMP molecules are needed to activate two catalytic subunits of protein kinase A, each active subunit phosphorylates hundreds or thousands of proteins before it returns to the regulatory subunits.
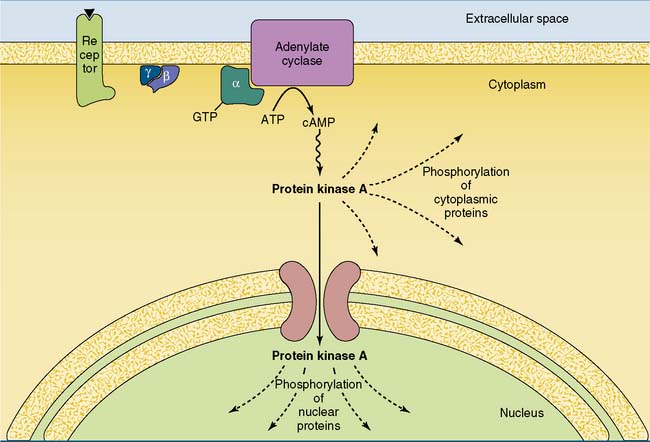
Some hormones do not stimulate but rather inhibit adenylate cyclase (see Table 17.1). In most cases inhibition is mediated by the αi subunit of an inhibitory G protein (Gi). Most cells possess both Gs-linked and Gi-linked hormone receptors, and the activity of adenylate cyclase depends on the balance between stimulatory and inhibitory hormones (Fig. 17.7).
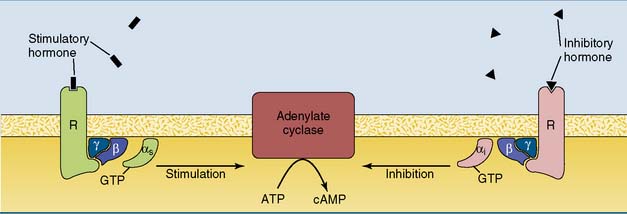
Many hormones and neurotransmitters can act through different receptors and second messengers. For example, epinephrine (adrenaline) can raise the cAMP level by activating β-adrenergic receptors, or it can reduce cAMP by activating α2-adrenergic receptors. A third receptor type, the α1-adrenergic receptor, raises the calcium level by activating the IP3 second messenger system (see section “Phospholipase C generates two second messengers”). Therefore the effect of epinephrine depends on the type of receptor that is present on the cell. Acetylcholine acts not only on nicotinic receptors, which are ligand-gated ion channels, but also on several subtypes of muscarinic receptors, which are linked to G proteins and either lower the cAMP level or raise the levels of IP3 and calcium.
CLINICAL EXAMPLE 17.5: Toxic Thyroid Nodules
Most cases of hyperthyroidism (“thyrotoxicosis”) are caused by Graves disease (see Chapter 16). Benign thyroid adenomas that overproduce the hormones (“toxic nodules”) are a less common cause of thyrotoxicosis. These tumors are derived from a single cell that proliferates abnormally as a consequence of somatic mutations.
CLINICAL EXAMPLE 17.6: Pseudohypoparathyroidism
The usual cause of pseudohypoparathyroidism is an abnormal Gs protein that couples poorly between the PTH receptor and adenylate cyclase. Many patients with this disorder have mild skeletal deformities, a condition known as Albright hereditary osteodystrophy. This condition is caused by a heterozygous gene defect inherited from the mother. The gene is paternally imprinted by DNA methylation (see Chapter 7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


